Около 40 % эпилептических припадков в первые годы жизни вызваны эпилептической энцефалопатией развития DEE) [1,2]. DEE - это группа заболеваний, характеризующихся эпилептическими припадками или эпилептиформной активностью, выраженной задержкой или регрессом психомоторного развития, а также когнитивными и поведенческими нарушениями [3]. Припадки и задержка развития при DEE имеют общую, обычно генетическую, этиологию, влияют друг на друга, но прогрессируют независимо. Часто эпилепсия начинается так рано, что невозможно определить ее основную причину. Таким образом, последствия для нейроразвития при DEE связаны с сочетанием прямого влияния генетического варианта и воздействия эпилептиформной активности, которые в той или иной степени могут вносить вклад в патогенез [4-7].
Как приступы, так и прогрессирование когнитивных нарушений приводят к тяжелым последствиям, снижая качество жизни и создавая финансовые и эмоциональные трудности для семьи. В зависимости от варианта DEE смертность в возрасте до 20 лет при некоторых синдромах может достигать 25 %, а остальные пациенты страдают от психических, поведенческих и двигательных расстройств [8-10]. Ситуация усугубляется ограниченной эффективностью существующей медикаментозной терапии в купировании приступов и улучшении неврологического состояния [9]. В наши дни ранняя диагностика является ключевым фактором в управлении исходом DEE - своевременное выявление заболевания и его этиологии напрямую коррелирует с более благоприятным исходом лечения и долгосрочным прогнозом [5].
Патогенные варианты встречаются у 30-50% пациентов с DEE [11-13]. Технологии секвенирования нового поколения значительно ускорили выявление генетических изменений у пациентов с DEE [14]. Идентификация и характеристика таких вариантов позволяет понять молекулярные механизмы заболевания. Выявление основных механизмов может стать основой для персонализированной терапии, которая не только облегчит тяжесть приступов, но и улучшит когнитивные показатели детей, страдающих этим заболеванием [15]. Не менее важно понять генетическую этиологию и выявить корреляции между генотипом и фенотипом, чтобы облегчить диагностику и консультирование семей пациентов [16,17].
В последнее десятилетие в литературе, посвященной DEE основное внимание уделялось каналопатиям. Хотя каналопатии действительно составляют значительную часть случаев DEE с патогенными вариантами генов, выявлено множество других, ассоциированных с DEE эти варианты приводят к нарушению различных аспектов развития и функционирования мозга, таких как метаболизм, пролиферация предшественников, миграция нейронов, формирование дендритов и аксонов и синаптогенез. Подавляющее большинство обзоров по DEE посвящено эпилепсии и энцефалопатии при DEE и зачастую не обсуждают механизмы когнитивных нарушений. Однако интеллектуальная недостаточность не менее тяжела для пациентов и усложняет возможное лечение. Поэтому важно рассматривать механизмы возникновения припадков и задержки развития в сочетании друг с другом. Цель данного обзора - оценить последние публикации о не каналопатиях при DEE с акцентом на механизмы, связывающие эпилепсию с умственной отсталостью.
2. Pathogenesis of Developmental Delay and Intellectual Disability in Developmental and Epileptic Encephalopathy
Для эпилептических энцефалопатий (DEE) характерны такие сопутствующие неврологические патологии, как задержка развития и умственная отсталость. Когнитивные нарушения при DEE обычно диагностируются в младенчестве или раннем детстве [18]. Когнитивные нарушения при DEE являются следствием как основной энцефалопатии, так и сопутствующих припадков или эпилептиформной активности, выявляемой на EEG [4,5]. Длительное перевозбуждение нейронов во время припадков, независимо от пути их возникновения, способствует снижению когнитивных функций [19-21].
В норме когнитивные функции зависят от слаженной работы нейронных сетей, обеспечивающих эффективную передачу информации между различными областями мозга. При эпилептических энцефалопатиях судороги и эпилептиформная активность приводят к хаотичным разрядам, которые нарушают эту координацию и разрушают функциональные связи между нейронами [22]. В результате этого процесса нарушается интеграция сенсорной информации, исполнительный контроль и память. Это впоследствии нарушает развитие когнитивных способностей. Хронические нарушения нейронной синхронности усугубляют задержку развития и способствуют формированию стойкой умственной отсталости [23].
Патогенные варианты генов в большинстве случаев являются основной причиной умственной отсталости и задержки развития. Многие из этих вариантов связаны с дисфункцией ионных каналов, связанных с напряжением. Поскольку ионные каналы влияют на генерацию, распространение и контроль потенциалов действия, такие изменения часто приводят и к эпилептической активности [6,24]. Нарушение ионного баланса вызывает перевозбуждение нейронов, что приводит к искажению нейронных функций и впоследствии к когнитивным нарушениям [25-28]. Для большинства DEE характерно раннее начало. Нервная система наиболее уязвима к аномальной электрической активности в период раннего развития. Поэтому дисфункции ионных каналов способствуют повреждению или гибели нейронов, что еще больше усугубляет когнитивные нарушения и отклонения в развитии [21].
Кроме того, в некоторых исследованиях описаны нарушения синаптической пластичности из-за патогенных вариантов генов [29]. Действительно, пластичность нейронов является ключевым механизмом, лежащим в основе высших когнитивных способностей, таких как память и обучение [30]. При эпилептических энцефалопатиях изменения в экспрессии генов, регулирующих синаптическую пластичность, могут усугублять когнитивные нарушения, поскольку нейроны теряют способность адаптироваться к новым условиям или формировать новые связи [31]. Это еще раз подчеркивает сложность и многофакторность когнитивных нарушений при DEE, поскольку в патогенез DEE могут быть одновременно вовлечены ионные каналы, синаптическая пластичность и нейронные сети в целом [32].
Метаболические нарушения в нейронах приводят к тяжелым последствиям из-за чрезмерного накопления продуктов метаболизма, нарушения энергетического обмена и снижения торможения. Энергетический дисбаланс и накопленные метаболиты нарушают сигнализацию между клетками, способствуют воспалению нейронов или даже приводят к гибели нейронов. Эпилептическая активность нарушает метаболизм как в очаге припадка, так и в соседних областях [33].
Повышенная возбудимость может нарушать миграцию нейронов во время развития. Было показано, что временная активация мигрирующих проекционных нейронов (PNs) в развивающейся коре головного мозга вызывает изменения в транскрипции метаботропных глутаматных рецепторов, преждевременное дендритное ветвление и удержание нейронов в более глубоких слоях коры [6,34]. С другой стороны, гиперполяризация предшественников нейронов в желудочковой зоне неокортекса мыши вызывала изменения транскрипции и характеристик клеточного деления на более поздних стадиях развития: они приобретали необычные морфологические и молекулярные особенности. С другой стороны, промежуточные предшественники, экспрессирующие транскрипционный фактор Tbr2, формировались преждевременно. Все это указывает на то, что изменения биоэлектрической активности в ходе нейрогенеза могут нарушать временные программы дифференцировки нейронов, вызывая аномальные функции нейронов [35].
Эпилептические припадки и эпилептиформная активность повреждают нейронные сети, которые являются основным субстратом когнитивных функций. Основой функционирования нейронных сетей в коре головного мозга и гиппокампе является долговременное потенцирование (LTP) - процесс усиления проводимости нервных импульсов при синаптической передаче в течение длительного периода времени [36]. Он играет важную роль в обучении, памяти и развитии сенсорных систем. LTP отвечает за стабильную работу и укрепление синаптических связей [37]. Хронические припадки, в свою очередь, приводят к нарушению LTP [18,38].
Место эпилептической активности в мозге является решающим фактором для когнитивного исхода припадков - повреждение функциональных зон приводит к их нарушению. Гиппокамп считается одной из наиболее важных структур в формировании памяти. После припадков пирамидальные клетки гиппокампа образуют аномальные нейронные связи, что приводит к нарушению долгосрочной, краткосрочной и пространственной памяти [21,39]. Часто повреждается и лобная доля неокортекса, что приводит к нарушению логического мышления, рабочей памяти, контроля эмоций и волевых движений [20].
Фенотипический спектр вариантов генов, вызывающих DEE, очень широк. Различные варианты нарушений в одном гене могут вызывать различные последствия для белка: Варианты с «усилением функции» чаще всего приводят к раннему развитию DEE, а варианты с «потерей функции» - к позднему развитию DEE, умственной отсталости и ASD [40]. В этом обзоре мы сосредоточимся на трех основных механизмах умственной отсталости при DEE (рис. 1). Кроме того, мы опишем новые гены, вовлеченные в патогенные молекулярные каскады и обойденные вниманием в других обзорах, посвященных ASD.
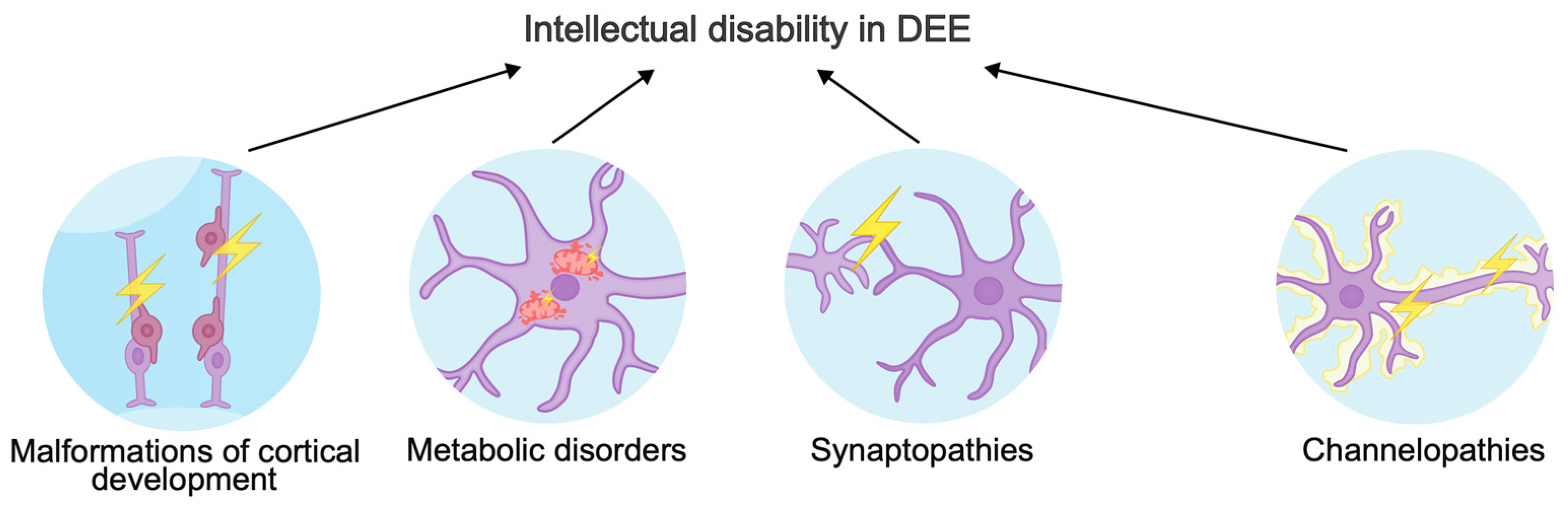
Рисунок 1.
Механизмы, лежащие в основе умственной отсталости при энцефалопатиях развития и эпилептических энцефалопатиях.
3. Molecular Mechanisms Underlying Developmental and Epileptic Encephalopathy
Наиболее частые причины DEE имеют генетическую этиологию [10,17]. Заболевания часто являются моногенными, но встречаются и олигогенные варианты [41,42]. По данным секвенирования экзома и анализа всего генома, основными причинами являются варианты
de novo, но существуют и другие наследственные формы: аутосомно-рецессивные, доминантные и Х-сцепленные варианты [12,42-44]. Большинство патогенных вариантов связано с каналопатиями, метаболическими нарушениями, мембранным транспортом, а также ростом и пролиферацией предшественников в процессе нейрогенеза [45]. Краткое описание генов представлено ниже в таблице 1.
Таблица 1. Патогенные варианты, ассоциированные с умственной отсталостью при энцефалопатиях развития и эпилептических энцефалопатиях.
3.1. Malformations of Cortical Development as a Cause of DEE
Развитие коры головного мозга основано на правильной временной активации и инактивации жестко регулируемых генетических программ, которые контролируют пролиферацию и дифференцировку нейронных предшественников, спецификацию, миграцию и формирование нейронных цепей. Все это определяет формирование правильно функционирующего мозга. Аномалии развития коры головного мозга (MCDs) связаны с патогенными вариантами генов, нарушающих эти процессы, это приводят к аномалиям морфологии и функций мозга. Патогенные варианты могут быть связаны с генами, кодирующими модификаторы хроматина, факторы транскрипции и РНК-связывающие белки, которые контролируют процесс нейрогенеза. Мутации в таких генах вызывают нарушения нейрального развития, включая DEE [75-81] (рис. 2).

Рисунок 2.
Патогенные варианты генов приводят к нарушению ключевых процессов нейрогенеза, что может быть причиной умственной отсталости при DEE. Гены разделены на подгруппы по механизму, исходя из их роли в патогенезе умственной отсталости при DEE.
3.1.1. Neuronal Progenitor Proliferation Disruption
Описаны патогенные варианты нейрон-специфического комплекса ремоделирования хроматина (BAF), который регулирует экспрессию генов, участвующих в контроле ламинирования неокортикальных клеток, ветвления дендритов и формирования синапсов [82-85]. Патогенные варианты BAF, ACTL6B, ассоциированы с тяжелыми формами DEE с глубокой задержкой развития и умственной отсталостью [86-90]. Белок ACTL6B контролирует доступность транскрипции ДНК и необходим для поддержания баланса пролиферации клеток-предшественников нейронов (NPC). Пролиферативное состояние поддерживается комплексом (np)BAF с ACTL6A во время нейрогенеза, в то время как дифференцировка NPCs в зрелые постмитотические нейроны требует переключения с ACTL6A на ACTL6B в комплексе (n)BAF [83,89,91,92]. Наиболее распространенные патогенные варианты обнаружены в актин-подобных доменах белка, что приводит к потере функции белка и нарушению сборки комплекса BAF. Они вызывают дисрегуляцию генов, связанных с самообновлением нейронных предшественников, что приводит к аномальной цитоархитектуре неокортекса и впоследствии к умственной отсталости. Так, среди клинических признаков вариантов ACTL6B - умственная отсталость, задержка развития, отсутствие речи, гипомиелинизация, агенезия мозолистого тела и тяжелая эпилепсия [86,88,89,93-96]. Эксперименты с культурой клеток нейронов продемонстрировали нарушение формирования синапсов, что подтверждает важное влияние ACTL6B на развитие нейронов [89]. Таким образом, потеря функции ACTL6B снижает способность нейронов формировать синаптические связи и приводит к нарушению дифференцировки нейронов, что играет критическую роль в патологии DEE и умственной отсталости.
Патогенные варианты INPP4A связаны с нарушением внутриклеточных сигнальных путей. Биаллельные усеченные варианты вызывают спектр нарушений нейроразвития от легкой умственной отсталости до DEE и микроцефалии [56]. Ген INPP4A кодирует фермент инозитолполифосфат-4-фосфатазу, который участвует в метаболизме инозитола в фосфоинозитидных сигнальных путях и регулирует транспорт везикул, что имеет решающее значение для функционирования нейронов. Мышиные модели демонстрируют повышенную гибель нейронов из-за дефекта пролиферации [97-100]. Более того, у мышей с патогенным вариантом Inpp4a наблюдаются дефекты в развитии стриатума, что важно для нормального моторного и когнитивного поведения. Кроме того, в культурах нейронов было показано, что INPP4A регулирует синаптическую локализацию NMDAR, защищает нейроны от excitotoxic гибели и тем самым поддерживает функциональную целостность мозга [101]. Таким образом, INPP4A критически важен для развития нервной системы, поскольку продукт этого гена влияет на функционирование множества сигнальных путей, поддерживает клеточный гомеостаз и нейрогенез, а также играет роль в клеточной пролиферации и подавлении глутаматной эксайтотоксичности [101-104].
Следующий интересный игрок в нарушениях нейрогенеза - SMC1A. Этот ген кодирует компонент когезинового комплекса, который участвует в сегрегации хромосом во время репликации, репарации ДНК и регуляции транскрипции [105-108]. Патогенные варианты этого гена могут приводить к синдрому Cornelia de Lange со специфической задержкой развития, а также вызывать раннюю DEE. Более того, DEE, ассоциированное с SMC1A, характеризуется глобальной задержкой развития и встречается исключительно у женщин, что связано с вероятной мужской летальностью [106-110].
Изменения этого гена в эмбриональных стволовых клетках мозга приводили к уменьшению количества петель ДНК, потере когезина на промоторах и энхансерах, изменению экспрессии генов и дефектам пролиферации. Предположительно, дефекты SMC1A приводят к хромосомной нестабильности и нарушениям экспрессии генов на ранних стадиях развития мозга, способствуя возникновению патологий нейроразвития [105-108,111].
Нарушение нейрогенеза может быть вызвано и другими механизмами. Например, формирование правильного пула изоформ мРНК необходимо для выхода нейрональных предшественников из клеточного цикла. Нарушение программ сплайсинга РНК в период раннего развития мозга играет важную роль в этиологии NDDs [112-115]. Ген GEMIN5 кодирует многофункциональный белок, участвующий в сборке малых ядерных рибонуклеопротеинов (snRNPs), регуляции сплайсинга пре-мРНК и, в целом, в трансляции [116-124]. Дефекты GEMIN5 ассоциируются с атрофией мозжечка, умственной отсталостью, двигательными нарушениями и эпилептической энцефалопатией раннего детского возраста (EIDEE) [3,125,126]. Патогенные варианты GEMIN5 нарушают способность GEMIN5 взаимодействовать с другими белками комплекса SMN или связывать snRNA. «Loss-of-function» варианты GEMIN5 являются наиболее распространенными и приводят к нарушению трансляции и снижению связывания внутреннего сайта входа в рибосому (IRES), что вызывает дефекты в экспрессии генов, необходимых для развития нервной системы [122,124,127-129]. «Loss-of-function» варианты GEMIN5 повышают активность путей, связанных с сигнализацией постсинаптической мембраны и с секрецией нейротрансмиттеров, и снижают активность путей, связанных с развитием клеток, внеклеточного матрикса и ядерного транспорта [129]. Считается, что Gemin5 играет критическую роль в раннем развитии млекопитающих. Гомозиготные нокаутные модели являются эмбрионально летальными [130,131]. Известно, что биаллельные варианты GEMIN5 также вызывают задержку развития, двигательную дисфункцию и атрофию мозжечка. Вероятно, это связано со снижением уровня белков сборки snRNP-комплекса и дефектами регуляции РНК-мишени [129].
Ген HNRNPU - один из генов, регулирующих процессинг РНК. Он кодирует гетерогенный ядерный рибонуклеопротеин U (hnRNP U) - белок, играющий ключевую роль в поддержании трехмерной структуры генома [81,132-138]. HNRNPU широко экспрессируется в мозге, особенно в коре, гиппокампе и мозжечке [139]. Патогенные варианты признаны причинами NDDs умственной отсталости, аутизма и ранней DEE (EIEE54) [55,114,115,140,141]. HNRNPU-ассоциированные патологии развития в основном обусловлены дефектами с потерей функции, которые приводят к спектру нейрональных патологий: аномальной миграции нейронов, увеличению боковых желудочков и дефектам формирования мозолистого тела [81,142-144]. Мышиные модели с гаплонедостаточностью Hnrnpu демонстрируют аномалии в организации мозга и патологии путей проекции и миграции нейронов. Поскольку все зарегистрированные человеческие варианты гетерозиготны, гомозиготные HNRNPU, вероятно, приводят к пренатальной смерти у людей, как и у мышей [80,81,115,137,145]. Гаплонедостаточность HNRNPU предположительно не позволяет нейрональным предшественникам выйти из клеточного цикла и начать дифференцировку, нарушая траекторию развития нейронов. Это приводит к нарушению нейронального развития и вызывает целый спектр неврологических расстройств [115,146,147].
3.1.2. Neuronal Differentiation Disruption
Другим геном, вовлеченным в DEE и имеющим важное значение для нейрогенеза, является CUX2 [148]. CUX2 кодирует фактор транскрипции, регулирующий пролиферацию нейрональных предшественников в субвентрикулярной зоне (SVZ), их дифференцировку и выход из клеточного цикла. CUX2 экспрессируется в конце клеточного цикла, перед окончательным митозом нейронных предшественников в SVZ [149-151]. Cux-2 и Cux-1 вместе являются ранними маркерами дифференцировки нейронов: ген Cux1 участвует в пролиферации, а ген Cux2 контролирует спецификацию типов клеток и дифференцировку нейронов. Также известно, что экспрессия гена Cux необходима для дифференцировки и развития интернейронов [149-154]. Задержка экспрессии CUX2 может приводить к аномальному завершению клеточного цикла, вызывая дефекты кортикогенеза и последующие патологии нейроразвития [149-151].
Другая группа генов, патогенные варианты которых вызывают умственную отсталость, вовлечена во внутриклеточные сигнальные каскады. Так, в последние годы были выявлены de novo варианты субъединиц G-белков. Например, патогенные варианты гена GNAO1 ассоциированы с тяжелыми неврологическими синдромами, начиная от задержки развития с двигательными нарушениями и заканчивая EIEE [155-162]. GNAO1 кодирует альфа-субъединицу Gα, гетеротримерного G-белка, который регулирует внутриклеточную сигнализацию. Самый высокий уровень GNAO1 наблюдается в конусе роста дифференцирующихся нейронов. Gα отвечает за молекулярную сигнализацию, которая направляет навигацию конуса роста под действием внешних сигналов. Этот процесс является ключевым для правильного формирования нейронной цепи [162-165]. Дефекты Gα нарушают способность белка связывать и гидролизовать GTP, снижают взаимодействие с белками-партнерами и приводят к потере белка в цитоплазматической мембране. Из-за ключевой роли в многочисленных сигнальных системах нейронов варианты Gα вызывают различные дефекты развития. Например, они приводят к нарушению роста и удлинения нейритов [166-170]. Мышиные модели Gnao1 демонстрируют раннюю постнатальную летальность, уменьшенное количество предшественников кортикальных нейронов и увеличенные боковые желудочки [171]. У пациентов с нарушенным GNAO1, напротив, наблюдалось снижение уровня генов нейрогенеза, повышенная экспрессия маркеров астроцитов, дефекты дифференцировки и аномальное формирование нейронных сетей. У них наблюдались низкие концентрации внутриклеточного свободного кальция и нарушенная реактивность нейротрансмиттеров. Таким образом, патогенные варианты GNAO1 нарушают нейронную коммуникацию [172].
Следующий пример - ген SP9, который кодирует транскрипционный фактор семейства Sp/KLF, необходимый для регуляции экспрессии генов в нейрогенезе. SP9 экспрессируется в ходе эмбриогенеза в коре головного мозга и базальных ганглиях, где он необходим для правильной дифференцировки, миграции нейронов и формирования нейронных цепей [59,173]. В нескольких исследованиях сообщалось о двух основных типах NDDs вызванных дефектами в гене SP9. Потеря функции третьего C2H2-связывающего домена приводит к задержке развития, эпилепсии и аутистическим расстройствам, в то время как изменения во втором домене приводят к EE [59,174]. SP9 участвует в развитии кортикоспинального тракта и тангенциальной миграции GABA-ергических нейронов. Ген также играет важную роль в пролиферации и дифференцировке striatopallidal проекционных нейронов. Без SP9 кортикальные интернейроны не мигрируют в кору или стриатум. У модельных животных, нокаутированных по Sp9, наблюдается снижение плотности корковых интернейронов, аномальная организация сети и дефектный рост аксонов. Таким образом, нокаут Sp9 приводит к когнитивным и двигательным нарушениям, сходным с теми, что наблюдаются у пациентов с DEE [59]. По-видимому, потеря функции SP9 нарушает транскрипционный контроль генов, критически важных для кортикогенеза, что приводит к неправильной локализации нейронов, дефектному формированию цепей и изменению синаптической пластичности [59,175].
3.1.3. Neuronal Migration Disorders
Нарушение миграции нейронов во время развития мозга может быть причиной DEE [160,176]. Соответствующая регуляция динамики цитоскелета, в частности микротрубочек, необходима для миграции нейронов [177]. Тубулины играют важную роль в этом процессе, будучи необходимыми для митоза, аксонального транспорта, миграции нейронов и формирования синапсов [178-180]. Один из этих генов, TUBA1A, кодирует изотип α-тубулина, который высоко экспрессируется в постмитотических клетках нейронов, но отсутствует в нейронах-предшественниках [181-184]. α-тубулин образует гетеродимеры с β-тубулином, формируя полимеры микротрубочек. Дисфункция микротрубочек может приводить к различным нарушениям нейронального развития, называемым тубулинопатиями [180,185,186].
Патогенные варианты TUBA1A являются основной генетической причиной лиссэнцефалии, а также могут приводить к микроцефалии, аномалиям мозолистого тела, гетеротипии серого вещества и DEE [61,187-189]. Варианты, вызывающие потерю функции (LoF) TUBA1A, приводят к недостатку тубулина в клетках, поскольку эти варианты не способны полимеризовать микротрубочки. С другой стороны, варианты с усилением функции (GoF) способны формировать микротрубочки, но не могут взаимодействовать с динеином [180,185,190,191].
У мутантов Tuba1a нарушена радиальная миграция нейронов. Мышиные модели патогенного варианта Tuba1a демонстрируют перинатальную смертность в гомозиготном состоянии и тяжелые пороки развития мозга к E16.5. У этих мышей наблюдается уменьшение толщины слоев CTIP2+ и PAX6+ нейронов и апоптотическая гибель нейронов. Тяжелый фенотип нейроразвития связан с уменьшением количества постмитотических и апикальных предшественников нейронов [180,192,193].
Другим ключевым геном для миграции нейронов является DCX. Он кодирует белок дублкортин, который участвует в организации микротрубочек во время дифференцировки нейронов и миграции интернейронов в кору головного мозга [194-199]. Патогенные варианты DCX нарушают структуру N- и C-концевых областей белка, которые необходимы для связывания с микротрубочками и неполимеризованным тубулином. Эти изменения в белке DCX препятствуют нормальному взаимодействию нейронов, что приводит к нарушению миграции нейронов и дефектам в архитектуре развивающейся коры головного мозга [195,200,201].
Эти патогенные варианты клинически ассоциируются с тяжелыми пороками развития мозга, гетеротопией подкорковых полос, лиссэнцефалией, умственной отсталостью, эпилепсией и DEE [195,197,202]. Наиболее тяжелые варианты фенотипа связаны со сдвигом рамки считывания de novo, в то время как миссенс-варианты вызывают более легкие дефекты развития. DCX расположен на Х-хромосоме. Поэтому наиболее тяжелые последствия патогенных вариантов этого гена наблюдаются у мужчин, проявляясь в виде тяжелой MCD, лиссэнцефалии, задержки развития, умственной отсталости и судорог. У женщин, напротив, наблюдается более мягкий фенотип в виде гетеротопии [195,203-205].
3.1.4. Dendrito- and Axonogenesis Disorders
Морфогенез нейронов, включающий формирование дендритных разветвлений и аксонов, зависит от действия множества молекул, контролирующих структуру и поддержание цитоскелета. Одним из таких факторов является ген CYFIP2, который играет важную роль в регуляции актинового цитоскелета через комплекс WAVE [206]. Когда малая Rho ГТФаза Rac1 связывается с белком CYFIP2, активируется WAVE-комплекс, который взаимодействует с Arp2/3 [207]. Это взаимодействие способствует полимеризации актиновых филаментов и поддерживает динамику полимеризации/деполимеризации, необходимую для роста и ветвления нейритов [208]. Дефекты в CYFIP2 нарушают этот процесс, что приводит к дестабилизации актиновых филаментов и нарушению роста [209]. Это проявляется в снижении способности нейронов формировать листовидные ламеллиподии и синаптические контакты, что влечет за собой дефекты синаптической пластичности и нарушение миграции нейронов [210]. У пациентов патогенный вариант CYFIP2 приводит к тяжелой DEE, психомоторной задержке, интеллектуальным нарушениям, гипотонии и поведенческим расстройствам и может быть связан с синдромом хрупкой Х [211,212].
SPTAN1 - еще один ген, важный для поддержания структурной целостности нейронов. SPTAN1 кодирует белок Spectrin αII, который также участвует в организации актина и стабилизации структуры мембраны. SPTAN1 обеспечивает структурную целостность цитоскелета и нормальное функционирование нейронов [213]. Спектрин II связывается с актиновыми филаментами, формируя опорную сеть под клеточной мембраной, что важно для поддержания механической стабильности мембран и синаптической пластичности [60]. Этот белок также необходим для сборки узлов Ранвье [214]. Патогенные варианты SPTAN1 приводят к дефектам аксонов и нарушению клеточной архитектуры, что приводит к эпилепсии, задержке развития, ASD, микроцефалии, спастической параплегии и синдрому West [213,215,216].
Динамика цитоскелета также регулируется геном RHOBTB2, который кодирует белок семейства ГТФаз Rho-типа. RHOBTB2 участвует в регуляции цитоскелетной динамики, клеточной миграции и везикулярного транспорта, влияет на клеточную дифференцировку и апоптоз [217]. Взаимодействие RhoBTB с белком Cullin3, входящим в состав убиквитин-протеасомного комплекса, может регулировать уровни специфических белков, необходимых для нормального развития дендритов и синаптической пластичности. В контексте RHOBTB2 ассоциация с Cullin3 предполагает, что миссенс-варианты могут нарушать механизм деградации, влияя на стабильность белков, необходимых для нормального развития дендритов и функционирования нейронов. RHOBTB2 играет важную роль в контроле клеточного цикла, участвуя в регуляции клеточной дифференцировки и апоптоза [218,219]. Нокаут RhoBTB в дендритах нейронов дрозофилы выявил критическую роль в формировании дендритной архитектуры, уменьшая количество дендритных отростков. Миссенс-варианты в кодирующей области BTB-домена RHOBTB2 ассоциированы с DEE, что указывает на их важность в развитии нейронов и, возможно, в регуляции дендритогенеза [220]. Однако точные молекулярные механизмы, связывающие миссенс-варианты с развитием нейронов, остаются малоизученными, что требует дальнейших исследований для характеристики их роли в невропатологии.
Ген DYNC1H1 также регулирует функции цитоскелета. Он содержит цитоплазматическую тяжелую цепь динеина, которая опосредует связывание динеиновых комплексов с микротрубочками [221]. Этот процесс критически важен для поддержания гомеостаза нейронов и доставки ключевых компонентов, участвующих в синаптической активности, таких как рецепторы нейротрансмиттеров, предшественники синаптических пузырьков и другие [222,223]. Нарушения функции DYNC1H1 могут приводить к дефектам сворачивания белков и связывания микротрубочек [224]. У пациентов с патологическими вариантами DYNC1H1 наблюдаются задержка нейроразвития, DEE и, в некоторых случаях, аномальная морфология мозга, включая микроцефалию и другие фенотипы [221].
Нарушение функции тормозных нейронов при DEE может быть связано со снижением уровня белка Caspr2, кодируемого геном CNTNAP2 [225]. Этот ген кодирует контактин-ассоциированный белок-подобный 2, член семейства нейрексинов - молекул клеточной адгезии, участвующих в формировании синаптических контактов [226,227]. CNTNAP2 необходим для миелинизации, направления аксонов, организации дендритных разветвлений и формирования корешков, а значит, контролирует формирование нейронных сетей в целом [228]. Дефицит CNTNAP2 вызывает повышенную возбудимость нейронов [229]. В частности, рецессивные варианты гена CNTNAP2 влияют на уровень и функции GluA1, субъединицы AMPA-рецепторов, регулирующих возбуждающую синаптическую передачу [230]. Нарушение работы CNTNAP2 приводит к изменению экспрессии, поверхностной локализации и эндоцитоза GluA1, ослабляя синаптическую пластичность и модулируя активность кальций-зависимых сигнальных путей [231]. У пациентов с рецессивным вариантом гена CNTNAP2 наблюдаются когнитивные нарушения, языковые расстройства, судороги и синдром фокальной эпилепсии с кортикальной дисплазией (CDFE), а также снижение числа GABA-ергических интернейронов и нарушения миграции нейронов, что указывает на глубокие дефекты формирования и функционирования нейронных сетей [47].
Ген EEF1A2 играет важную роль в трансляции и организации цитоскелета нейронов. Он кодирует эукариотический фактор элонгации трансляции 1А2, который влияет на процесс синтеза белка. EEF1A2 связывается с аминокислотами и тРНК и участвует в переносе тРНК на А-сайт рибосомы, что необходимо для удлинения полипептидной цепи в процессе трансляции. Патогенные варианты EEF1A2 ассоциированы с DEE, задержкой развития и микроцефалией [232], поскольку они нарушают трансляцию (из-за повышенного связывания тРНК), снижая скорость трансляции. Это влияет на морфологическое развитие нейронов коры головного мозга. Патогенный EEF1A2 обладает более низкой актин-связывающей активностью. Таким образом, EEF1A2 выполняет две функции: регуляция трансляции и организация цитоскелета нейронов [233]. При нокауте EEF1A2 в клетках глиобластомы человека нарушался процесс пролиферации и дифференцировки клеток [234].
Таким образом, в процессе развития коры головного мозга множество молекулярных путей взаимодействуют между собой, создавая сложную нейронную сеть, составляющую кору головного мозга. Нарушение любого из этих путей может привести к серьезным патологическим состояниям. Это, в свою очередь, может привести к развитию интенсивной и иногда многоочаговой эпилептической активности, связанной с DEE.
3.2. Synaptopathies—Synaptic Transmission Disorders
Патогенные варианты генов, влияющие на пре- и постсинаптические трансмембранные белки, могут приводить к DEE как напрямую, так и опосредованно. Пре- и постсинаптические мембраны участвуют в транспорте синаптических везикул в аксонах и в инициации потенциала действия в дендритах (рис. 3).
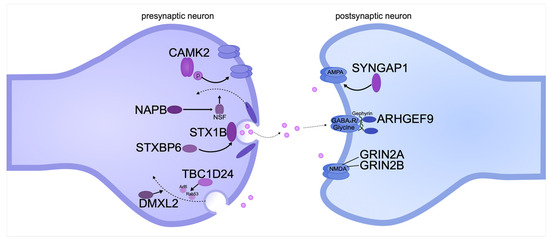
Figure 3.
Pathogenic gene variants encoding pre- and postsynaptic transmembrane proteins can cause intellectual disability in DEE. The diagram shows the location of proteins relative to the synaptic cleft and the functions they perform. The proteins are systematized based on recent publications on DEE.
Белковый комплекс SNARE играет важную роль в пресинаптической мембране [235]. Одним из членов комплекса является синтаксин-1B, который кодируется геном STX1B. Основная роль этого белка - прикрепление синаптических везикул к пресинаптической мембране [236]. Синтаксин-1B имеет две конформации: открытую, которая необходима для формирования комплекса SNARE, и закрытую, которая инициирует реакцию слияния везикул [237,238]. De novo патогенные варианты STX1B клинически ассоциируются с DEE и генерализованной эпилепсией с фебрильными судорогами [67]. Чаще всего миссенс-варианты представляют собой варианты с потерей функции в открытой конформации белка, что приводит к нарушению сборки комплекса и транспорта везикул. Патогенные варианты закрытой конформации белка приводят к нарушению меж-белковых взаимодействий и нормального слияния пресинаптических везикул [239]. У мышей с нокаутом гена Stx1b наблюдаются тяжелые судороги и преждевременная смерть, связанные с нарушением высвобождения нейротрансмиттеров в GABA- и глутаматергических синапсах [240]. Помимо STX1B, в поддержании верности процессов слияния везикул с мембраной участвуют и другие регуляторные белки, например продукт гена STXBP6. STXBP6, кодирующий синтаксин-связывающий белок 6, также известный как амизин, участвует в модуляции активности синтаксина и контроле мембранных взаимодействий, что необходимо для нормального функционирования синаптического аппарата, а именно движения нейронных везикул [241]. У пациента с эпилептической энцефалопатией и расстройством аутистического спектра (ASD) был обнаружен усеченный вариант белка, кодируемого геном STXBP6 [242]. У мышей с делецией этого гена была снижена масса тела, что также является одним из фенотипов у некоторых пациентов с ASD. Однако когнитивные навыки у этих мышей не были нарушены [243].
Высвобождение нейротрансмиттера происходит через Ca(2+)-индуцированное слияние синаптических везикул, опосредованное комплексом SNARE [244]. Комплекс SNARE связан с белком βSNAP, который является продуктом гена NAPB. βSNAP является одним из кофакторов NSF-АТФазы, которая необходима для синаптической передачи, поскольку этот фермент участвует в разборке и утилизации белков SNARE-комплекса [245]. Цельноэкзомное секвенирование у трех братьев и сестер с тяжелой умственной отсталостью и DEE выявило делецию семи пар оснований в гене NAPB, что привело к усечению белка на 46 %. У детей развились эпилептические припадки в возрасте до 6 месяцев и тяжелая регрессия развития к 2 годам [246]. Недавно было проведено полноэкзомное секвенирование арабо-палестинской семьи, состоящей из трех однояйцевых близнецов с диагнозом «синдром Cohen». Близнецы страдали ранней эпилептической энцефалопатией, аутизмом и умственной отсталостью. Анализ данных секвенирования выявил патогенный вариант, затрагивающий сайт сплайсинга гена NAPB [247]. У мышей со сниженной экспрессией βSnap наблюдались эпилептические припадки, затем атаксия и, в некоторых случаях, смерть [245].
Среди белков постсинаптической мембраны патогенные варианты SYNGAP1 наиболее часто ассоциируются с DEE [248]. Этот белок является ключевым посредником в сигнальном каскаде RAS, активируемом NMDA-рецептором. Во время LTP SYNGAP1 активирует RAS-GTPase (SynGAP) в глутаматергических нейронах, что приводит к встраиванию AMPA-рецепторов и увеличению площади синаптической поверхности [249-251]. Патогенные варианты SYNGAP1 влияют на глутаматергические синапсы и усиливают активность глутаматных рецепторов, повышая вероятность эпилептогенеза [252]. мРНК SYNGAP1 имеет несколько альтернативно сплайсированных вариантов, кодирующих различные изоформы белка, которые отличаются по структуре, функции и временной экспрессии. Идентифицированы четыре С-концевые изоформы: α1, α2, β и γ. Изоформа β экспрессируется в начале постнатального развития, в то время как α2 экспрессируется на более высоком уровне в зрелом мозге [253,254]. Это объясняет различия в тяжести фенотипа; например, нонсенс-опосредованный распад, вызванный дефектами в ранних изоформах, приводит к полной потере генного продукта. Более мягкий исход SYNGAP1-DEE наблюдается у пациентов с вариантами сайтов сплайсинга в экзонах с 1 по 4 [68].
Еще одним геном, ассоциированным с синаптопатией, является TBC1D24. Этот ген кодирует белок, активирующий малые ГТФазы Arf6 и Rab35, которые действуют антагонистически. Они необходимы для мембранного транспорта в синапсах, а также между плазматической мембраной и эндоцитарными компартментами [255]. TBC1D24 имеет широкий спектр экспрессии и обнаруживается во всех слоях коры головного мозга и гиппокампа. Патогенные варианты TBC1D24 вызывают дисрегуляцию синаптических везикул, что приводит к избыточной нейротрансмиссии. Кроме того, он нарушает нормальное удаление дефектных белков через эндосомные пути, что приводит к их накоплению и дисфункции нейронов. Эти изменения способствуют развитию широкого спектра эпилептических фенотипов и других нарушений нейроразвития у пациентов [69]. Нокаут гена Tbc1d24 в первичных кортикальных нейронах крысы выявил нарушение формирования начальных сегментов аксонов и возбудимости нейронов. Этот фенотип был связан с повышенной активацией ГТФазы Arf6, которая необходима для спецификации аксонов и удлинения нейритов [256].
Ген DMXL2 кодирует крупный белок, который связан с везикулярным транспортом и играет ключевую роль в регуляции синаптической передачи. Нарушение функции белка DMXL2 может приводить к нарушениям синаптического эндоцитоза и рециркуляции везикул. Это связано с тем, что DMXL2 регулирует закисление внутриклеточных компартментов через вакуолярный протонный насос (V-ATPase) [257]. Кроме того, белок DMXL2 действует как модулятор сигнального пути Notch и необходим для рекрутирования в хроматин Notch-зависимых факторов транскрипции [258]. Патогенные варианты DMXL2 могут нарушать эти процессы, что приводит к дисбалансу возбуждения/торможения в нервной системе, вызывая повышенную возбудимость нейронов. Это, в свою очередь, ассоциируется с развитием эпилептических припадков и тяжелой задержкой развития, характерной для DEE [6]. Повышенная возбудимость нейронов, лежащая в основе DEE также может быть связана с дисфункцией глутаматных рецепторов. В частности, гены GRIN2A и GRIN2B кодируют субъединицы NMDA (N-метил-D-аспартат) рецепторов, которые являются подтипами глутаматных рецепторов. Они играют ключевую роль в синаптической пластичности, обучении и памяти. Эти рецепторы контролируют поступление ионов кальция, натрия и калия через мембрану нейрона, что необходимо для передачи возбуждающих сигналов в мозге [65,259]. Повышенная активность NMDA-рецепторов приводит к чрезмерному притоку кальция в клетки, что может вызвать гиперактивность нейронов и, как следствие, их гибель [260]. Существует множество редких вариантов генов GRIN2A и GRIN2B, ассоциированных с неврологическими заболеваниями. В настоящее время в GRIN2A зарегистрировано 304 варианта, вызывающих DEE а в GRIN2B (ClinVar) - 273 варианта, вызывающих DEE [261]. Фенотипические проявления в этих генах были подробно изучены в ряде клинических исследований. У пациентов с патогенными вариантами GRIN2A/GRIN2B наблюдаются тяжелые формы эпилептической энцефалопатии, сопровождающиеся задержкой моторного и когнитивного развития. Эти клинические проявления коррелируют с нарушениями, наблюдаемыми на уровне синаптической передачи и активности нейронов [262].
Ген ARHGEF9 кодирует белок collibostin (Cb), который регулирует динамику актинового цитоскелета и синаптическую активность через активацию Rho ГТФаз, в частности Cdc42 [263]. Он напрямую взаимодействует с каркасным белком gephyrin и необходим для формирования gephyrin-зависимых кластеров GABA А на постсинаптической мембране [264]. Взаимодействие с Cb происходит благодаря наличию домена SH3, который связывается с большой внутриклеточной петлей ?2 субъединицы рецепторов GABA А [265]. Точечные мутации в ARHGEF9 нарушают тормозную синаптическую передачу за счет взаимодействия с GABA- и глициновыми рецепторами, что приводит к повышенной возбудимости нейронов и когнитивным нарушениям. Это связано с развитием эпилепсии, аутизма, умственной отсталости и, в некоторых случаях, определенных лицевых дисморфий [62,147].
Ca 2+/кальмодулин-зависимая протеинкиназа II (CAMK2) - один из важнейших ферментов, участвующих в синаптической пластичности и формировании памяти [266]. Белок состоит из двух преобладающих субъединиц - альфа (CAMK2A) и бета (CAMK2B), которые в значительной степени гомологичны друг другу и, вероятно, могут замещать функции друг друга при инактивации одной из них [267]. Патогенные варианты CAMK2A или CAMK2B вызывают умственную отсталость, аутизм и DEE у людей [268]. Фермент CAMK2 является частью Ca-зависимого сигнального пути и фосфорилирует различные субстраты, ответственные за LTP [269]. При активации CAMK2A оказывает значительное влияние на дендритные отростки и постсинаптическую плотность, взаимодействуя с белками, связанными с ферментом, в частности с субъединицами GluN2B NMDA-рецепторов [270]. При нарушении участка аутофосфорилирования CaMK2A у мышей наблюдаются дефекты пространственного обучения и памяти [271].
Таким образом, синаптопатии являются одним из ключевых механизмов, лежащих в основе эпилептических энцефалопатий и нарушений нервного развития в целом. Нарушения синаптической пластичности приводят к дисрегуляции нейронных связей, что вызывает эпилептическую активность в мозге и значительные когнитивные и моторные нарушения. Это, несомненно, подчеркивает важность изучения синаптопатий для понимания патогенеза эпилептических расстройств и связанных с ними задержек развития [31].
3.3. Metabolic Disorders
Мозг млекопитающих отличается высокой энергетической потребностью. Большая часть энергии расходуется на активацию потенциалов действия и синаптическую передачу. Она обеспечивается за счет гликолиза и митохондриального дыхания. С другой стороны, потребность в энергии во время нейрогенеза также чрезвычайно высока. Поэтому неудивительно, что аномальная биоэнергетика и дисфункция митохондрий в нейронах вызывают когнитивные расстройства [272].
Одно из таких расстройств вызывается патогенными вариантами HK1, который кодирует гексокиназу HK1. Этот фермент осуществляет АТФ-зависимое фосфорилирование глюкозы до глюкозо-6-фосфата (Г6Ф) в процессе гликолиза [273]. HK1 экспрессируется преимущественно в нейронах и астроцитах мозга, а дисфункция гена связана с многочисленными нарушениями развития, включая нейроразвивающие расстройства (NDDs) и DEE [72,273].
Патогенные варианты
HK1 могут вызывать умственную отсталость через несколько механизмов, связанных с важнейшими функциями гексокиназы в клеточном метаболизме и деятельности нейронов. HK1 состоит из двух симметричных мономеров, содержащих связанный с альфа-спиралью регуляторный N-концевой домен и каталитический C-концевой домен [274,275]. Продукт фосфорилирования этой гексокиназы, G6P, связывается с обоими доменами фермента, что приводит к конкурентному ингибированию связывания АТФ и ингибированию киназной активности [276,277]. Этот процесс нарушается при наличии миссенс-вариантов в альфа-спирали и регуляторном домене фермента, что делает невозможным связывание G6P с доменами HK1, и фермент теряет способность к саморегуляции. Такие дефекты приводят к «усилению функции» HK1: фермент продолжает конститутивно фосфорилировать глюкозу, что приводит к накоплению метаболитов и повреждению митохондрий [278]. Предполагается, что это приводит к накоплению неправильно сформованных белков, стрессу эндоплазматического ретикулума, дисфункции митохондрий, апоптозу и гибели клеток [72,279]. Это может привести к потере нейронов в областях мозга, отвечающих за обучение, память и познание, таких как гиппокамп и префронтальная кора. Патогенные варианты
HK1 снижают доступность энергии в клетках мозга, а дефицит энергии приводит к нарушению активности нейронов. Это, в свою очередь, может привести к дефектам в формировании нейронных сетей в критические периоды развития и, как следствие, к когнитивным нарушениям и умственной отсталости [6,72] (рис. 4).
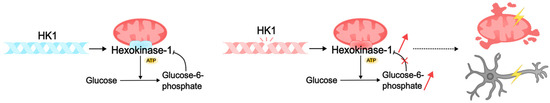
Figure 4.
Pathogenic variants of HK1 can lead to intellectual disability by disrupting cellular metabolism. Normally, HK1 hexokinase catalyzes the phosphorylation of glucose to glucose-6-phosphate (G6P), which binds to the enzyme domains. Competitive inhibition with ATP blocks kinase activity. Pathogenic HK1 disrupts the reverse binding of G6P to the enzyme domains. As a result, the kinase continues to constitutively phosphorylate glucose, which leads to the accumulation of metabolites, damage to mitochondria, and death of neurons. Lack of energy and dysfunction of neural networks can subsequently lead to intellectual disability in DEE.
Другие патогенные варианты, например, вызывающие дисфункцию мембранных транспортеров, также могут приводить к нарушению биоэнергетики нейронов. Ген SLC25A12 кодирует митохондриальный аспартат-глутаматный транспортер (AGC1/Aralar), компонент малат-аспартатного челнока (MAS), экспрессирующегося в основном в нервной системе и мышцах. Этот транспортер осуществляет антипорт (antiport) цитозольного глутамата и протонов в обмен на интрамитохондриальный аспартат. Функция MAS необходима для поддержания окислительно-восстановительного баланса между цитозольным гликолизом и митохондриальным дыханием и обеспечивает синтез АТФ, что важно для нейронов, которые имеют высокие энергетические потребности [280-283]. Патогенные варианты SLC25A12 приводят к дефициту AGC1, который вызывает детскую эпилептическую энцефалопатию с глобальной задержкой психомоторного развития и гипомиелинизацией мозга [284]. Патогенные варианты приводят к нарушению стробирования транспортера, что ограничивает конформационные изменения белка для высвобождения субстрата в митохондриальном матриксе и нарушает метаболическую активность клеток [285-288]. Исследования на мышиных моделях Agc1-knockout демонстрируют снижение клеточного дыхания в мозге, уменьшение уровня аспартата и нарушение метаболизма глутамата. В результате нейроны, лишенные AGC1, не способны поддерживать нормальную метаболическую активность [282,288]. AGC1 играет центральную роль в биоэнергетике нейронов, а поскольку рост и дифференцировка нейронов требуют повышенного производства энергии, этот белок очень важен в процессе нейрогенеза [272,289].
Кроме того, патологии AGC1 в нервной системе приводят к дефициту аспартата и ограниченному биосинтезу N-ацетиласпартата (NAA), который необходим для синтеза миелина. Снижение миелинизации нарушает нормальное развитие аксонов и их способность передавать сигналы, вызывая патологию нейротрансмиссии, в частности глутаматергической, что объясняет интеллектуальный дефицит, наблюдаемый у пациентов [70,290-292] (рис. 5).
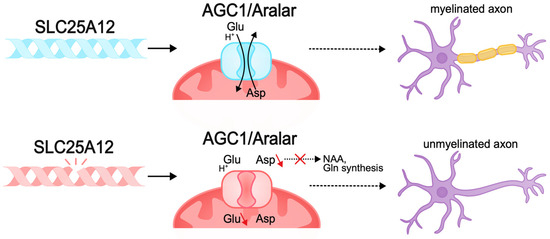
Рисунок 5.
Патогенные варианты SLC25A12 могут приводить к нарушению биоэнергетики нейронов и аксональной миелинизации. Ген SLC25A12 кодирует митохондриальный аспартат-глутаматный транспортер (AGC1/Aralar). Патогенные варианты SLC25A12 приводят к нарушению функционирования ворот транспортера и неспособности антипорта аспартата и глутамата. Недостаток аспартата в нервной системе приводит к дефициту биосинтеза N-ацетиласпартата (NAA), который необходим для синтеза миелина и миелинизации аксонов. Снижение миелинизации нарушает нормальное развитие аксонов и их способность передавать сигналы, что может быть причиной умственной отсталости при DEE.
Мышиные модели патологий этого транспортера имеют выраженную гипомиелинизацию, а также нарушают развитие аксонов коры головного мозга и постнатальное развитие кортико-гиппокампальных нейронов [282,293-296]. Таким образом, патогенные варианты SLC25A12 приводят к нарушению функции нейронов и развитию тяжелых форм эпилептической энцефалопатии с сопутствующим интеллектуальным дефицитом, что связано с нарушениями кортикогенеза, миелинизации и глутаматергической передачи вследствие метаболических расстройств.
Известны случаи умственной отсталости, вызванной дефектами гликозилирования белков. Эта пост-трансляционная модификация белков играет важную роль во многих внутриклеточных процессах, в том числе в синаптической пластичности. На животных моделях нарушений гликозилирования в нервной системе наблюдаются нарушения синаптогенеза, аномалии развития гиппокампа и умственная отсталость [297-300]. Одним из генов, участвующих в контроле гликозилирования, является ALG13. Продукт этого гена участвует в пост-трансляционной модификации белков путем N-гликозилирования, а экспрессия гена наблюдается преимущественно в нейронах коры головного мозга и гиппокампа [301]. Патогенные варианты ALG13 приводят к врожденным нарушениям гликозилирования и DEE. Они характеризуются глобальной задержкой развития с регрессом, гипотонией и двигательными нарушениями. В основном они диагностируются у женщин [302-304]. Внутри клетки ALG13 образует гетеродимерный комплекс с белком ALG14, который выполняет вспомогательную функцию по прикреплению ALG13 к мембране эндоплазматического ретикулума. Вместе они образуют функциональную гликозилтрансферазу UDP-GlcNAc, которая переносит N-ацетилглюкозамин на аспарагиновые остатки белков [305-309]. Этот процесс необходим для правильного сворачивания белков и формирования функциональных гликопротеинов, что обеспечивает их стабильность, сортировку и транспорт, а также важен для осуществления межклеточных взаимодействий [301,310-312]. Белок ALG13 имеет несколько изоформ: длинную (ALG13-is1) и короткую (ALG13is2). Эти изоформы идентичны в каталитическом домене N-концевой области, но значительно отличаются в С-концевой области - части белка, отвечающей за транспорт белков в эндоплазматический ретикулум. Патологии вызываются мутациями как в каталитическом, так и в С-концевом доменах, которые нарушают активность белка, его взаимодействие с мембраной эндоплазматического ретикулума и способность гликозилировать белки. Известно, что дефекты в С-концевой области длинной изоформы ALG13 приводят к развитию и эпилептической энцефалопатии, умственной отсталости и нарушениям гликозилирования I типа [308,313-315]. У мышиной модели Alg13KO наблюдаются когнитивные нарушения, снижение сложности и длины дендритов, а также плотности дендритных отростков в гиппокампе. Вероятно, снижение когнитивных способностей при патологии ALG13 обусловлено неспособностью формировать правильные синаптические связи [316-319]. Кроме того, было установлено, что потеря ALG13 характеризуется гибелью нейронов и реактивным астроглиозом и может снижать тормозную синаптическую передачу путем регуляции транскрипции субъединицы GABA A R α2, что усугубляет патологии синаптической пластичности [301]. В совокупности патогенные варианты ALG13 вызывают глубокие когнитивные нарушения, задержку развития и тяжелый фенотип DEE, обусловленный нарушениями гликозилирования и пластичности нейронов.
Другими примерами нарушений гликозилирования являются патогенные варианты ST3GAL3, который кодирует трансмембранный фермент Гольджи sigleosyltransferase ST3Gal-III. Этот фермент катализирует перенос сиаловой кислоты на галактозу в ганглиозидах и гликопротеинах. Сиалогликаны критически важны для нервной системы, поскольку они необходимы для нормальной работы нейронов, межклеточной коммуникации, миелинизации и синаптической пластичности [320-324]. Нарушения в работе фермента ST3Gal-III приводят к снижению уровня сиалогликанов, что нарушает функции нервной системы, влияет на когнитивное развитие и способность к обучению. Дефицит сиалогликанов нарушает стабильность и функции мембранных белков, что препятствует нормальной передаче сигналов в нейронах [325,326].
Варианты потери функции ST3GAL3 приводят к синдрому West, синдрому DEE с регрессом развития и умственной отсталостью, и тяжелой несиндромальной аутосомно-рецессивной умственной отсталости (NSARID) [324-329].
Исследования на моделях мышей
St3gal3-null и St3gal2/3-null показали, что нарушение работы генов приводит к недостатку сиалилирования гликопротеинов и, как следствие, к гипомиелинизации, нарушению пролиферации олигодендроцитов и аномальному формированию узлов Ранвье. Подобно человеческому фенотипу, у этих мышей наблюдались серьезные когнитивные нарушения, снижение двигательной координации и гиперактивное поведение. Кроме того, отсутствие адекватного сиалилирования нарушало правильное функционирование синапсов, что приводило к снижению синаптической пластичности и ухудшению обучения и памяти. В целом, патогенные варианты
ST3GAL3 приводят к дефициту гликозилирования ганглиозидов и гликопротеинов, а затем и к когнитивной дисфункции, и у пациентов с патогенным
ST3GAL3 наблюдается тяжелая умственная отсталость, задержка развития и DEE [323,325] (рис. 6).
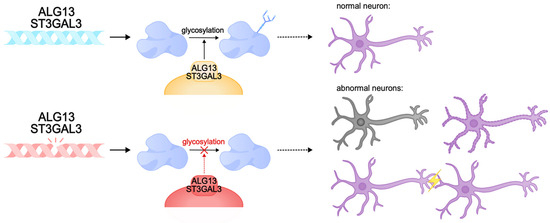
Рисунок 6.
Figure 6. Pathogenic variants of ALG13 and ST3GAL3 can lead to congenital glycosylation disorders. These genes encode transmembrane glycosylation enzymes that play a critical role in the nervous system. Pathogenic variants can cause abnormalities in synapse function, neuronal membrane formation, and neuronal death. These abnormalities can result in intellectual disability, delayed development, and DEE.
Таким образом, метаболические нарушения в развивающейся нервной системе являются одним из механизмов развития умственной отсталости при DEE. Среди рассмотренных нами причин патогенные варианты некоторых ферментов и транспортеров вызывают дефекты биоэнергетики и пост-трансляционной модификации, что приводит к патологии синаптической передачи и миелинизации, патологии формирования нейронной сети и гибели нейронов. В конечном итоге все это приводит к нарушению межнейронной коммуникации и последующей умственной отсталости.
4. Conclusions
Эпилептические энцефалопатии (DEEs) - это группа заболеваний, характеризующихся эпилептическими припадками, эпилептиформной активностью между приступами и тяжелой задержкой развития с когнитивными нарушениями. Эти патологии часто имеют общую этиологию и влияют друг на друга, но развиваются параллельно и по-разному.
В основе DEE часто лежит генетическая этиология. В последнее десятилетие, благодаря развитию секвенирования нового поколения, многие исследовательские группы по всему миру обнаружили множество патогенных вариантов, вызывающих DEEs. Часто это моногенные расстройства, которые либо возникают de novo, либо наследуются рецессивно. Наиболее частые варианты, вызывающие DEEs, связанные с каналопатиями, нарушают функцию генов, кодирующих вольтаж-зависимые натриевые и калиевые каналы, такие как, например, SCN2A и KCNQ2.
Однако в последнее время было описано множество вариантов, вызывающих DEEs, продукты генов которых контролируют другие процессы, помимо проводимости тока: метаболические нарушения, мембранный транспорт, рост и пролиферацию в ходе нейрогенеза. Эти данные показывают, что патогенез DEE выходит далеко за рамки нейронной передачи, и любое нарушение правильного количества и соотношения различных типов нейронов, их расположения, синаптического входа и выхода, аксонального и дендритного транспорта и потребления энергии может нарушить правильный баланс возбуждения/торможения и вызвать очень тяжелые последствия в работе мозга, которые будут проявляться в эпилептиформной активности и приводить к умственной отсталости.
Выявление и детальное исследование генетических причин DEE и вовлеченных в них молекулярных каскадов важно для понимания молекулярных основ патогенеза, ответственных за возникновение этих расстройств. Понимание этих путей и определение корреляции между генотипом и фенотипом может помочь в диагностике и генетическом консультировании семей пациентов. Хотя во многих случаях к моменту диагностики заболевания цитоархитектура мозга уже окончательно деформирована и лечение невозможно, существуют случаи DEEs, когда структура мозга не претерпевает значительных изменений. Такие случаи потенциально могут лечиться индивидуально, в зависимости от молекулярного каскада, на который влияет патогенный вариант гена. Например, если причиной является нарушение обмена веществ, может быть использована заместительная терапия в сочетании с генной терапией. В мозг доставляются генные конструкции или мРНК, которые заменяют сбойные белки. С другой стороны, если основной причиной заболевания является «усиление функции» определенного гена, можно подобрать небольшую молекулу-ингибитор, которая ослабит гиперактивность генного продукта. Общей чертой и проблемой многих DEE является фармакорезистентность к противоэпилептическим препаратам. В этом случае в отдельных случаях можно использовать животные модели, воспроизводящие патологию, чтобы подобрать лечение с помощью комбинации противоэпилептических препаратов.
Раскрытие молекулярных механизмов патогенеза умственной отсталости при DEE может стать основой для персонализированной терапии, которая позволит улучшить не только тяжесть приступов, но и когнитивные результаты у пострадавших детей.
