Название «амиотрофический» состоит из трех греческих слов. «А» — отрицание, «мио» — относящийся к мышцам, «трофика» — питание. Таким образом, мышцы у больных БАС испытывают дефицит питательных веществ, а потому атрофируются. Слово «боковой» указывает на область спинного мозга, в которой располагаются погибшие участки нервных клеток. С дегенерацией данной области происходит ее уплотнение («склероз» как раз и означает «затвердевание»). Возможно, самой ужасной особенностью заболевания является то, что больной не утрачивает умственных способностей и осознает все, что с ним происходит.
Наследственная форма БАС встречается лишь у 5—10% больных. Ранние проявления заболевания у разных пациентов могут быть неодинаковы, но обычно все начинается со слабости в руках и ногах, речевых расстройств, мышечных судорог, ограниченности движений. Как только затрагиваются дыхательные мышцы, больного помещают в стационар и переводят на искусственную вентиляцию легких.
При БАС поражаются только двигательные нейроны, но мотонейроны, ответственные за произвольные движения глаз и мочеиспускание, по неизвестным причинам долгое время остаются интактными. Администрация по контролю над пищевыми продуктами и лекарственными средствами США (РАО) одобрила применение лишь одного препарата для лечения БАС. Это рилузол, вещество, продлевающее жизнь на несколько месяцев; скорее всего его действие состоит в подавлении образования вредных химических соединений, повреждающих двигательные нейроны.
В 1993 г. обнаружен ген, ассоциированный с одной из форм наследственного БАС. Он кодирует фермент супероксид-дисмутазу (8SOD1), защищающий клетки от свободных радикалов (высоко реакционноспособных веществ, образующихся, в частности, в ходе нормальных метаболических процессов. Затем было идентифицировано более 100 мутаций в гене фермента, приводящих к развитию БАС. В последнее время появился целый ряд свидетельств в пользу того, что мутации в гене SOD1 обусловливают появление у кодируемого им фермента деструктивных свойств.
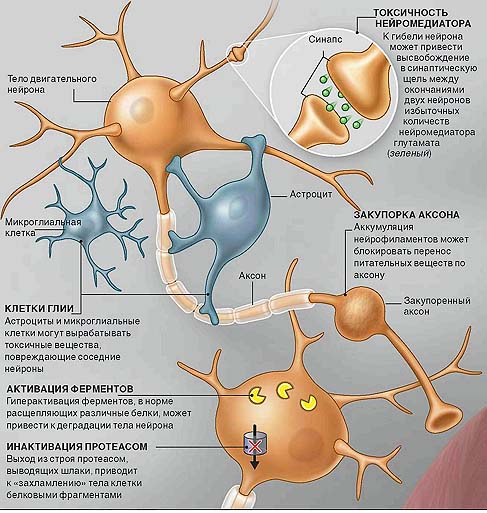
Становится все более очевидно, что дегенерация двигательных нейронов протекает по особому механизму. У этих нервных клеток необычайно длинный аксон. Его связь с мышцами устанавливается с помощью высвобождаемых в синаптическую щель веществ — нейромедиаторов. В случае БАС отмирание нервных клеток начинается в области синапсов, поэтому все усилия ученых направлены на поиски средств, которые могли бы защитить от разрушения именно эти структуры, а не только тела клеток.
Двигательные нероны можно подразделить на две группы: одни при БАС фрагментируются настолько, что их связь с мышцами полностью утрачивается, другие пытаются образовать новые ответвления. Выяснив, почему нейронам второй группы удается компенсировать дефект и в результате выжить, можно будет попытаться найти новые способы борьбы с БАС.
В аксоне в большом количестве представлены так называемые нейрофиламенты — белковые образования, отвечающие за сохранность его структуры. Показано, что при аномально высоком содержании нейрофиламентов в аксоне блокируется транспорт питательных и других веществ от тела нейрона к синапсу. Нарушение транспортных процессов, по-видимому, приводит к гибели тела двигательного нейрона. Мутациями в гене, кодирующем белки нейрофиламентов, обусловливается примерно 1% всех случаев заболевания БАС. В теле двигательных нейронов должно вырабатываться огромное количество энергии, которая обеспечивала бы функционирование чрезвычайно длинного аксона и синаптических окончаний. Образующиеся здесь митохондрии («энергетические фабрики» клеток) транспортируются вниз по аксону, а такие важные вещества, как факторы роста, поступают от периферической части нейрона к его телу. Поломка в любом звене транспортной системы может привести к сбою в работе двигательного нейрона.
Показано, что интактные глиальные клетки способствуют восстановлению поврежденных двигательных нейронов, и наоборот — если глиальные клетки поражены, они вызывают дегенерацию здоровых двигательных нейронов. Таким образом, очевидно, что в развитии БАС участвуют оба типа клеток.
Что там, за горизонтом?
Есть ли надежда, что когда-нибудь будут найдены эффективные способы лечения больных боковым амиотрофическим склерозом? В настоящее время уже идентифицирован ряд веществ, способных защищать от повреждений аксоны двигательных нейронов. Среди них — белок под названием цилиарный нейронный фактор, который поддерживает в жизнеспособном состоянии как двигательные, так и сенсорные нейроны. Далее, показано, что глиальные клетки вырабатывают нейротропное вещество, предотвращающее саморазрушение тел нейронов, но не влияющее на жизнеспособность аксонов.
В поисках молекул, защищающих аксоны, выявлено слияние двух разных генов,совместно кодирующих химерный белок, один из фрагментов которого необходим для эффективной работы клеточной системы выведения шлаков и функционирования фермента, который ускоряет синтез никотинамидаденин динуклеотида (НАД), участвующего во многих метаболических процессах в клетке. У мышей
NES8 аксоны поврежденных нервных клеток выходят из строя гораздо медленнее, чем у обычных животных.
| |
|
ПРОНИКНУТЬ В БОЛЬНОЙ МОЗГ
Не дожидаясь, пока будут найдены эффективные способы лечения БАС, исследователи и инженеры занимаются разработкой электронных устройств, способных регистрировать сигналы, которые посылает мозг парализованного больного. Такие устройства позволяют пациентам общаться с окружающими и управлять вспомогательными приспособлениями. Одни из так называемых интерфейсов мозг-компьютер (brain-computer interface, BCI) предполагают хирургическую имплантацию электродов, которые улавливают сигналы, исходящие от небольших групп нейтронов в двигательной области коры головного мозга. Другие используют электроды, фиксируемые на черепе, которые регистрируют волны электрической активности от миллионов нервных клеток. Нейрофизиологи Джонатан Вулпо (Jonathan Wolpaw), Тереза Вон (Theresa Vaughan) и Эрик Селлерз (Eric Sellers) из Центра Уодсуорта при Департаменте здравоохранения штата Нью-Йорк в Олбани создали BCI специально для страдающих БАС, который улавливает сигналы, возникающие в тот момент, когда больной сосредоточивает внимание на каком-либо объекте. Пациенту предъявляют таблицу из 72 букв, чисел, знаков пунктуации и других символов на экране компьютера. Как только высвечивается тот символ, на который направлено внимание больного, его мозг посылает специфические сигналы, а компьютер обрабатывает их и определяет, что именно он хочет «сказать»
Ингрид Уикелгрен, штатный редактор Scientific American
|
Исследуя поведение нейронов грызунов в культуре, Милбрант показал, что валлерова мутация приводит к повышению активности фермента, участвующего в синтезе НАД; концентрация последнего увеличивается и защита аксона становится более эффективной. В ходе дальнейших экспериментов ученый продемонстрировал, что при повышении уровня НАД происходит ускорение биохимических реакций, по-видимому связанных с продолжительностью жизни. Более того, на эти процессы влияют такие низкомолекулярные вещества, как резвератрол, содержащийся в кожице красного винограда, и эти же вещества замедляют разрушение поврежденных нейронов. Сегодня несколько фармацевтических компаний занимаются поисками соединений, которые могли бы останавливать развитие БАС и других нейродегенеративных заболеваний, ускоряя образование НАД.
Были получены трансгенные мыши, не способные вырабатывать фактор роста сосудистого эндотелия (ФРСЭ), белка, который участвует в регуляции роста кровеносных сосудов. Использовали для доставки целевого белка вырабатывающий его вирус. Вирусный препарат инъецировали в различные мышцы мышей, страдающих дефицитом ФРСЭ, откуда вирус проникал в нервные окончания двигательных нейронов, а затем — в тела клеток в спинном мозге (рис. вверху). Здесь он немедленно начинал синтезировать ФРСЭ в количествах, достаточных для того, чтобы прогрессирование БАС замедлилось. Более того было показано, что у людей, страдающих БАС, уровень ФРСЭ в крови существенно ниже, чем в норме. Сейчас Кармелит вместе со шведской фармацевтической компанией
Neuro/Nova готовится к проведению клинических испытаний препарата на основе ФРСЭ.
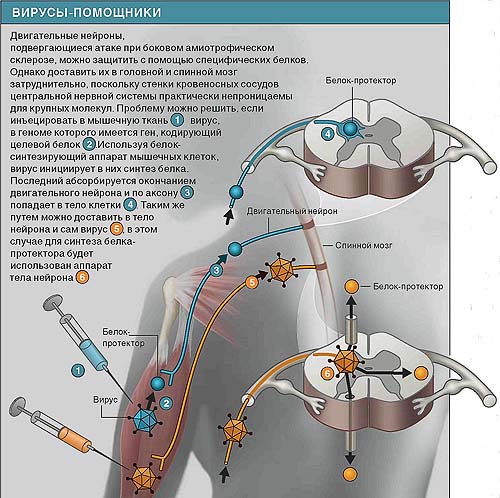
Еще один белок с протективными свойствами, на который возлагаются большие надежды, — инсулино-подобный фактор роста 1 (ИФР-1). Многообещающие результаты в отношении него получены в опытах с использованием вируса, вырабатывающего ИФР-1. От места инъекции в мышцах вирус переместился в окончания двигательных нейронов, а затем в тела клеток, где начался синтез ИФР-1. Продолжительность жизни животных увеличилась на 30% по сравнению с теми, кому вирус не вводили. В настоящее время идет подготовка к клиническим испытаниям, цель которых — проверить эффективность упомянутого метода доставки ИФР-1 на людях, больных БАС.
Одно из самых удивительных открытий последних лет состоит в том, что регулярные физические упражнения стимулируют рост новых нейронов, облегчают обучение, способствуют повышению уровня факторов роста в клетках нервной системы, а кроме того, защищают нейроны от посттравматических нарушений (последнее показано в опытах на животных). Возможно, такие занятия способствуют выработке ИФР-1, который повышает двигательную активность животных и даже восстанавливает поврежденные нейроны. Было решено попробовать совместить физические упражнения с введением в организм ИФР-1. Оказалось, что синергическое действие обоих факторов увеличивает продолжительность жизни грызунов примерно на 200 дней. Новые возможности лечения БАС появились и в связи с успехами,
| |
|
| ВОЗМОЖНЫЕ ПОДХОДЫ К ЛЕЧЕНИЮ
Исследователи рассматривают несколько стратегий борьбы с боковым амиотрофическим склерозом, которые могли бы замедлить прогрессирование болезни
НЕЙРОТРОПНЫЕ ФАКТОРЫ
По имеющимся данным, белками-протекторами могут служить такие вещества, как фактор роста сосудистого эндотелия и инсули-ноподобный фактор роста. Однако таким крупным молекулам трудно попасть из кровотока в центральную нервную систему, поэтому клиницисты рассматривают другой способ их доставки -с помощью вирусов, инъецируемых в мышечную ткань.
НЕБОЛЬШИЕ МОЛЕКУЛЫ
В кожице красного винограда содержится вещество под названием резвератрол. Он и сходные с ним соединения могут предотвращать дегенерацию нейронов, ускоряя синтез никотинамидаде-ниндинуклеотида (НАД), который участвует во многих клеточных метаболических процессах. Резвератрол без труда проходит через стенки кровеносных сосудов и попадает в ЦНС
СТВОЛОВЫЕ КЛЕТКИ
Трансплантированные стволовые клетки могут служить биологическим насосом, обеспечивающим поврежденные нейроны факторами роста. Как показали опыты на грызунах, стволовые клетки мигрируют к тем областям, где находятся поврежденные нейроны
РНК-ИНТЕРФЕРЕНЦИЯ
Синтетические фрагменты РНК связываются с комплементарными матричными РНК и блокируют синтез кодируемых ими токсичных белков в нейронах и глиаль-ных клетках
ФИЗИЧЕСКИЕ УПРАЖНЕНИЯ
Как показывают опыты на животных, больных БАС, у тех из них, кто имеет возможность «упражняться» на вращающемся колесе, прогрессирование заболевания замедляется. Особенно четко выражен этот эффект, если сопровождать упражнения введением инсулиноподоб-ного фактора роста
|
достигнутыми в последнее время в изучении стволовых клеток. Надежды возлагались на то, что в процессе дифференцировки они дадут начало новым нейронам, которые заменят поврежденные нервные клетки. Но последние исследования натолкнули на мысль, что пересаженные стволовые клетки могут играть роль насоса, снабжающего поврежденные нейроны факторами роста. Эксперименты на грызунах показали, что эти клетки на самом деле перемещаются от места введения к пораженным нейронам в ответ на сигналы, посылаемые последними. К дегенерации двигательных нейронов причастно также их окружение, поэтому лучше использовать трансплантанты, способные давать начало клеткам разного типа, а не только двигательным нейронам.
Новые перспективы открываются и в связи с обнаружением такого важного явления, как РНК-интерференция. В его основе лежит связывание коротких сегментов РНК со специфическими матричными РНК (мРНК), вследствие чего прекращается синтез кодируемых ими белков. Один из авторов статьи (Абишер) и сотрудники фирмы Oxford Biomedica использовали это явление для блокирования синтеза токсичного белка, введя в клетки-мишени SOD1-мышей вирус, кодирующий соответствующую РНКi. После данной процедуры прогрессирование БАС у животных замедлилось.
Результаты экспериментов подтолкнули исследователей к проведению клинических испытаний нового метода на больных наследственной формой БАС, несущих мутантный SOD1 ген. На начальных этапах предполагается введение синтетической РНКi непосредственно в спинномозговую жидкость пациентов. РНК сконструирована таким образом, чтобы ее связывание с молекулами мРНК произошло до того, как начнется синтез токсичного SOD1-белка в нейронах и глиальных клетках.
Клиническое использование любого из описанных выше подходов к лечению БАС сопряжено с большими трудностями. Прежде чем решиться на введение в организм человека факторов роста или РНКi с помощью вируса, необходимо убедиться в полной безопасности этой процедуры. Нужно оценить, насколько масштабным должно быть инъецирование, чтобы добиться успеха. Лучше всего вводить одновременно и факторы роста, и РНКi. Между тем некоммерческая организация ALS Association уже проводит клинические испытания комбинации препаратов, увеличивающих продолжительность жизни животных: целекоксиба (противовоспалительного средства, предотвращающее разрушение нервных клеток, обусловленное гиперактивностью глиальных клеток) и креатина.
Конечно, оптимальным выходом было бы предупреждение, а не лечение болезни. Путь к достижению этой цели мог бы включать, помимо всего прочего, физические упражнения и диету. Может быть, изменение образа жизни и кулинарных пристрастий поможет хотя бы снизить риск развития такого ужасного заболевания, как боковой амиотрофический склероз.
Перевод: Н.Н. Шафрановская
ДОПОЛНИТЕЛЬНАЯ ЛИТЕРАТУРА
|
| Unraveling the Mechanisms Involved in Motor Neuron Degeneration in ALS. Lucie Bruijn et al. in Annual Review of Neurosci-ence, Vol. 27, pages 723-749; July 2004.
Lentiviral-Mediated Silencing of SOD1 through RNA Interference Retards Disease Onset and Progression in a Mouse Model of ALS. Cedric Raoul et al. in Nature Medicine, Vol. 11, No. 4, pages 423-428; April 2005.
Silencing Mutant SOD1 Using RNAi Protects against Neuro degeneration and Extends Survival in an ALS Model. G. Scott Ralph et al. in Nature Medicine, Vol. 11, No. 4, pages 429-433; April 2005.
Axon Degeneration Mechnisms: Commonality amid Diversity Michael Coleman in Nature Reviews Neuroscience, Vol. 6, No. 11, pages 889-898; November 2005. |
Институт разработки терапии амиотрофического бокового склероза.
ALS Therapy Development Institute
ALS TDI институт и его ученые активно разрабатывают лечение для ALS. Это один из крупнейших некоммерческих биотехнологических институтов, сконцентрировавшийся на исследованиях ALS. Поддерживаемое ALS пациентами и их семьями, это благотворительное учреждение понимает насущные нужды по замедлению и остановке этой ужасной болезни. Базирующийся в Cambridge, MA, Институт является одним из лидеров по сбору данных и информации от академических и ALS исследовательских организаций и от пациентов и их семей (www.alstdi.org).
На рисунке представлены направления исследований Института
| |
|
Оксидативный стресс
Ученые ALS Therapy Development Institute тестируют указанные малые молекулы в попытке понять роль оксидативного стресса в клетках с ALS. |
|
SOD1 Misfolding
Superoxide Dismutase 1 (SOD1) участвует в очистке и превращении повреждающих супероксид свободных радикалов в менее вредные соединения. SOD1 преимущественно обнаруживается в цитоплазме, но также выявляется в митохондриях и клеточных ядрах. Обычно SOD1 образует гомодимер в 32 kd, образующий 8 ключевых beta цилиндров (barrel), которые связывают каталитический ион меди и структурный ион цинка. SOD1 является повсеместно встречающимся белком, но его клеточная концентрация существенно выше с метаболических тканях и ЦНС. Период полу-жизни белка SOD1 дикого типа большой, особенно в двигательных нейронах. Это увеличивает вероятность того, что белок SOD1 может быть подвержен пост-трансляционным модификациям в виде окисления. Полученные от человека моноклональные антитела, нацелены на неправильно упакованный SOD1, Neurimmune's NI-204 имеет целью снизить накопление неправильно упакованного SOD1 внутри двигательных нейронах при ALS. |
|
| |
Миелинирование
Тщательные исследования показали, что миелиновая оболочка аксонов двигательных нейронов нарушается. Исследовано несколько разных подходов по замедлению, остановке и устранении этих нарушений у пациентов ALS. |

| |
|
Оксидативный стресс< br />
Unfolded Protein Response
Считается, что при ALS накопление неправильно упакованных белков вызывает в нейронах стресс и активацию unfolded protein response (UPR) в качестве защитного механизма. При нормальных физиологических условиях система endoplasmic reticulum associated degradation (ERAD) в ER предназначена, чтобы устранять неправильно упакованные белки из ER в цитозоль для деградации посредством убиквитин протеосомной системы. Гомеостаз правильно упакованных белков в ER управляется десятками белков шаперонов и энзимов. Одним из наиболее многочисленных и критических ER шаперонов, поддерживающих этот гомеостаз, является шаперон BiP/Grp78.
BiP ассоциирует с тремя ER трансмембранными белками ATF6, Ire1 и PERK, которые обеспечивают определенные нижестоящие события реакции на неупакованные белки UPR. При нормальных физиологических условиях BiP ассоциирует с этими сигнальными молекулами, удерживая их прикрепленными к мембране ER но если концентрация неупакованных белков в ER увеличивается, то BiP отстраняется прочь от этих молекул, позволяя им отсоединяться от ER мембраны и обеспечивать нижестоящую передачу сигналов UPR.
Эти три регуляторных белка (ATF6, Ire1 и PERK) действуют совместно, чтобы снизить клеточные стрессы путем усиления аппарата белкового шаперона и путем снижения синтеза белка.
IRE1 это типа I ER трансмембранный белок с доменами цитоплазматической киназы и RNAse. Он образует олигомеры в условиях активации UPR , приводя к функциональной активации его функции RNAse. IRE1 гомо-олигомеры диссоциируют от плазматической мембраны ER в цитозоль. Активация IRE1's RNAse активности в цитозоле приводит к сплайсингу Xbp1 мРНК транскрипта на альтернативные сплайс-формы, которые распознаются аппаратом трансляции белка, который теперь транслирует Xbp1 в функциональный транскрипционный фактор, который проникает в ядро и изменяет экспрессию генов мишеней, ассоциированных с UPR.
PERK является типа I трансмембранного белка ER, чей цитозольный домен содержит киназный домен. В условиях ER стресса PERK отсоединяется от ER мембраны и его киназная активность фосфорилирует фактор инициации трансляции EIF2A. Это приводит к общему снижению трансляции белка в попытке ослабить накопление неправильно упакованных белков. Субнабор РНК преимущественно распознается с помощью фосфорилированного EIF2A фактора и они транслируются в функциональные белки. Сюда входят транскрипционные факторы CHOP и ATF4, а также GADD34, которые являются фосфатазами и осуществляют с помощью негативной петли обратной связи дефосфорилирование EIF2A. В условиях длительного ER стресса нижестоящие мишени транскрипционного фактора CHOP активируются и, являясь проапоптическими генами, приводят к гибели клетки.
ATF6 является типа II трансмембранным белком, который в условиях ER стресса транслоцируется в Гольджи, где отщепляется его N-конец, высвобождая активный транскрипционный фактор, который поступает в ядро, активируя ассоциированные с UPR гены.
Помимо своей роли в расщеплении Xbp1 продолжительная активация UPR приводит к IRE1 обусловленной активации ASK1 киназы. ASK1 является критическим компонентом двух путей клеточных стрессов: Jun N Terminal Kinase (JNK) Pathway и the p38 Map kinase pathway, которые активируют нижестоящие апоптические сигналы.
Модуляция этого критического пути обладает терапевтическим потенциалом у животных, моделирующих прионовую болезнь и болезнь Алцгеймера.
При ALS TDI исследует множественные пути, чтобы уменьшить поток апоптических сигналов, путем контроля долговременной активации UPR. Эти подходы сконцентрированы на малых молекулах ингибиторов критических регуляторов этого пути. |
|
Neuromuscular Junction
ALS является прогрессирующим нейромышечным заболеванием, характеризующимся мышечной атрофией и параличом. Характерной особенностью ALS является прогрессивная дегенерация нейромышечных соединений (NMJs).
У ALS пациентов было продемонстрировано, что время начала проявления симптомов соответствует более 50% потере NMJs в затрагиваемой группе мышц.
У взрослых моторные единицы иннервируются одиночным двигательным нейроном. Синаптические соединения в NMJ могут временно отсутствовать. Всё же система демонстрирует пластичность, делающую возможной повторную иннервацию предыдущим двигательным нейроном или даже соседними двигательными нейронами. Пока неясно, как поддерживается подобная пластичность и какие сигналы запускают процесс повторной иннервации.
Образование NMJs зависит от сложного сигнального каскада, который координирует образование зрелой синаптической щели между мышцами и нервными окончаниями. Ключевой передачей сигналов на мышечной (постсинаптической) стороне является рецепторная трозин киназа MuSK. Экспрессия MuSK предварительно закладывается в моторной единице и регулируется с помощью передачи сигналов Wnt и экспрессии MuSK лиганда LPR4. На стороне нерва (пресинаптической) экспрессия LRP4 рецептора Agrin входящими нервными окончаниями двигательного нейрона приводит к связыванию Agrin с LRP4, усиливая его ассоциацию с лигандом MuSK, приводя к фосфорилированию MuSK и образованию кластеров MuSK-LRP4-Agrin. Фосфорилирование MuSK приводит к внутриклеточному рекрутированию DOK-7. В присутствии Agrin Dok-7 становится фосфорилированным приводя к дальнейшему фосфорилированиюи MuSK и димеризации MuSK. Димеризация привлекает адапторные и закрепляющие белки Rho, Rac и rapysin, которые необходимы для образования с стабилизации NMJ. Рекрутирование адапторных белков вызывает доставку и образование кластеров ацетилхолиновых рецепторов. В результате образования кластеров ацетилхолиновых рецепторов происходит становление механизма высоко синхронизированного высвобождения нейротрансмиттеров в NMJ синапсах, которые превращают внутриклеточную передачу сигналов в электрический потенциал действия и в конечном счете в кинетические мышечные движения.
Пока неясна роль нарушений этих передач сигналов в повреждениях и нейродегенерации при ALS. Однако, некоторые нейротрофные факторы (IGF1, GDNF), микроРНК (mir-206) и происходящие из Шванновских клеток факторы (NOGO-A, Sema3A),по-видимому, играют как позитивную, так и негативную роль в репарации и стабилизации NMJ. ALS TDI обладает несколькими малыми молекулами и разрабатывает стратегии белковой терапии, нацеленные на эти ключевые медиаторы репарации NMJ в контексте нейродегенерации. |
|
| |
Immune Modulation
Недавние доказательства подтвердили, что ALS не является клеточно-автономным заболеванием, а сильно зависит от иммунной системы.
Описаны активация микроглии, астроцитоз и присутствие инфильтрующих воспалительных клеток с периферии. Происходит накопление IgG иммунореактивных отложений в спинном мозге при ALS, инфильтрация лимфоцитов, дендритных клеток, моноцитами и макрофагов в спинной мозг при ALS.
Общепринято, что два сигнала необходимы для максимальной активации T клеток. Привлечение T Cell Receptor (TCR) на T клетки с помощью антиген связывающего Major Histocompatibility Complex (MHC) на antigen presenting cells (APCs) и связывание CD28 на T клетках с B7 рецепторами на APCs.
Помимо B7 рецепторов член семейства Tumor Necrosis Factor (TNF) CD40 активируется на APCs (antigen presenting cells) после активации чужеродными антигенами. У нокаутных по CD40 мышей активация APCs с помощью чужеродного антигена активирует CD80, CD86 и MHC экспрессию на APCs, но неспособна активировать T клетки или вызывать гуморальный ответ. Связывание CD40 вызывает экспрессию адгезивных молекул и молекул с ко-стимулирующими эффектами на APCs, таких как CD44, ICAM1, ITGAX (CD11c ), CCL3 (Mip1a); Fcgr2b (CD32); ITGAM (CD11b); ITGB2 (LFA1/CD18) и MHC class II молекул.
Мы недавно охарактеризовали активацию ко-стимулирующего пути в спинном мозге, седалищном нерве и скелетных мышцах мышей SOD1G93A. Было установлено, что на периферии ко-стимуляторная активация ассоциирует с накоплением CD68+ моноцитов в седалищном нерве и с фокальной активацией в ЦНС. Активация ко-стимулирующих путей также обнаруживается в периферической крови у 47% проанализированных пациентов ALS. Воздействие anti-CD40L задерживало начало болезни, улучшало сохранение веса тела и удлиняло жизнь мышей, моделирующих ALS. Воздействие MR1 снижало количество CD68+ клеток в седалищном нерве за счет 42% снижения активации астроцитов в спинном мозге, улучшало жизнеспособность двигательных нейронов и снижало активацию ко-стимулирующего пути в спинном мозге и периферической крови.
Итак, имеются определенные доказательства, показывающие, что воспаление нейронов и нарушение гемато-энцефалического барьера связаны с прогрессированием болезни ALS. |
На странице этого сайта объявлено, что ALS Therapy Development Institute и Chaperone Therapeutics, Inc., приступают к совместной разработке лечения ALS. Chaperone Therapeutics, Inc. разработала несколько многообещающих малых молекул, вызывающих активацию HSF1, ключевого модулирующего пути, участвующего в укладке белков и защите от нейрональных стрессов.
Предполагается разработка базирующегося на клетках метода для тестирования способности шаперонов корректировать неправильно упакованные белки, защищать нейроны от индуцированной стрессами гибели клеток и метода оценки эффекта лечения в отношении прогрессирования ALS.
Терапия с помощью шаперонов усиливает естественную укладку клеточных белков и механизмы защиты от стрессов. Для получения большей информации обращайтесь www.chaperonetherapeutics.com.
Институт разработки терапии ALS помимо собственных исследований отслеживает разработки и клинические испытания препаратов других организаций.

Mitsubishi Tanabe Pharma Corporation объявила недавно, что она получла разрешение на продажу Radicut® (aka Edaravone or MCI-186) для лечения ALS в Японии. До этого использовани Radicut было ограничено из-за побочных эффектов в виде инсультов.
Согласно компании, Radicut очищает от свободных радикалов. Этот антиоксидантный подход обеспечивает защиту нервной системы.
Edison Pharmaceuticals, Inc. недавно объявила о клинических испытаниях при болезни Паркинсона и ALS одного и того же соединения, EPI-589. Предполагается измерять любые воздействие соединения на уровни антиоксиданта, наз. glutathione (GSH) в выборках крови и побочные эффекты.
Кстати не опубликовано данных относительно механизма действия EPI-589, иногда также обозначаемого (R)-troloxamide quinone. Нет публикаций и о преклинических испытаниях EPI-589 на моделях ALS. Однако, Edison Pharmaceuticals полагает, что уровни GSH будут модифицированы после лечения EPI-589. GSH присутствует почти во всех клетках и играет важную роль вместе с др. антиоксидантами в поддержании эффективного воздействия на реактивные виды кислорода.
Nicholas J. Kramer, Michael S. Haney, David W. Morgens,et al. CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nature Genetics, 2018; DOI: 10.1038/s41588-018-0070-7
ALS разрушает функцию мышц и нарушает способность головного мозга общаться с телом, упрощая произвольные мышечные движения, делая их затрудненными или даже невозможными. ALS характеризуется возникновением аномальных белковых глыбок в головном мозге. При ALS эти белковые глыбки или агрегаты является фатально токсичными для нейронов.
"Эти токсичные белковые агрегаты, скорее всего, и управляют патологическим процессом, но в действительности неизвестно, как они вызывают гибель нервных клеток," говорит Aaron Gitler.
Лаб. Gitler's и Bassik's использовали технологию CRISPR-Cas9 редактирования генов, чтобы проверить весь геном и найти гены, помогающие построить защиту от токсического белка.
A simple but deadly protein
Открытие, что мутации в гене C9orf72 являются довольно распространенной причиной, вызывающей ALS, помогло направить усилия на понимание, как ALS действует на молекулярном уровне. Мутантный ген C9orf72 содержит большой сегмент ДНК, который повторяет самого себя и когда часть гена ошибочно превращается в разные вредные белки, они тормозят функцию нейронов и приводят к гибели клеток.
"У здоровых лиц вы можете видеть 10-20 таких повторов ДНК," говорит Haney. "Но при ALS они увеличиваются до сотен и даже тысяч повторяющихся сегментов, и это становится матрицей для продукции этих токсических белков."
Удалось получить ответы на два основных вопроса: как токсические белки нарушают функцию во всем остальном здоровых нейронов? И существуют ли др. гены, которые прирожденно защищают от этих -- или напротив усиливают -- эффекты токсических белков в головном мозге?
Исследователи использовали геномный скрининг, при котором CRISPR-Cas9 конструкция меняла функцию каждого одиночного гена человека одновременно. В этом случае они использовали технологию продукции "генных нокаутов," целенаправленно воздействующие гены со свойствами молекулярных ножниц, которые осуществляли точные разрезы, делая их неспособными выполнять нормальную функцию.
Нокаут генов помог исследователям отметить гены, которые или усиливали токсичность, или предупреждали её: если вы идентифицировали ген и подвергали его нокауту и ALS белковые повторы переставали быть токсичными, то вы знали, что отсутствие этого гена в действительности защищает нейроны от дегенерации. И он может быть потенциальной мишенью для лекарственного воздействия.
Tmx2: A sentinel of cell death
После систематических нокаутов каждого гена в геноме человека и измерения токсичности ALS белков в клетках, исследователи нашли около 200 генов. которые будучи в нокауте или помогали защитить клетку от токсичных белков или делали её более чувствительной к ним. В конечном итоге меньшее количество генов было изучено с помощью двух последовательных нокаутных скринов в первичных нейронах мыши.
Была установлена горстка нокаутов, которые были особенно мощными защитниками. Один, напр., помогал блокировать критические входы, посредством которых токсические ALS белки проникали в клету и разрушали её. Но был и др. нокаут, в частности, который привлек внимание исследователей своей мистической способностью отвращать гибель нейронов. Ген обычно кодирует белок, наз. Tmx2, который обнаруживается в части клетки, наз. эндоплазматический ретикулум. Но когда он истощен в нейронах мышей in vitro, то клетки выживают практически 100% времени -- подобно прыжку, принимая во внимание, что доля выживания обычных нейронов составляет 10%.
"Мы предположили, что Tmx2 может стать прекрасной мишенью для поиска лекарств," сказал Haney. "Если мы имеем небольшую молекулу, которая может каким-то образом затруднять функцию Tmx2, то это может стать прорывом в терапии."
В данный момент роль Tmx2's в эндоплазматическом ретикулуме полностью ясна. Предполагается, что он участвует в реакции на разные внешне-средовые вещества. вызывающие стресс (stressors), особенно те, которые запускают клеточную гибель. Согласно полученным данным он может модулировать др. гены, которые запускают процесс клеточной гибели.
"Мы всё ещё находимся на ранней фазе, но я полагают выяснение в точности, что делает Tmx2 в норме в клетках станет прекрасным местом для старта -- чтобы показать, какая функция нарушается, когда эти токсические виды убивают клетку и это позволит найти пути, которые необходимо обнаружить," сказал Kramer.
Gitler и Bassik в настоящее время пытаются использовать тот же подход, чтобы выяснить дополнительные причины, вызывающие ALS, а также др. нейрологические болезни -- Huntington's, Parkinson's и Alzheimer's -- при которых действуют токсические белки. "Я полагаю, что это реально существующее приложение по CRISPR скринингу и это действует," говорит Bassik.