Посещений: 
БОЛЕЗНЬ ПОМПЕ
Генотерапия
Gene Therapy Developments for Pompe Disease Zeenath Unnisa, John K. Yoon,
Jeffrey W. Schindler et al.
 Biomedicines 2022, 10(2), 302; https://doi.org/10.3390/biomedicines10020302
|
Pompe disease is an inherited neuromuscular disorder caused by deficiency of the lysosomal enzyme acid alpha-glucosidase (GAA). The most severe form is infantile-onset Pompe disease, presenting shortly after birth with symptoms of cardiomyopathy, respiratory failure and skeletal muscle weakness. Late-onset Pompe disease is characterized by a slower disease progression, primarily affecting skeletal muscles. Despite recent advancements in enzyme replacement therapy management several limitations remain using this therapeutic approach, including risks of immunogenicity complications, inability to penetrate CNS tissue, and the need for life-long therapy. The next wave of promising single therapy interventions involves gene therapies, which are entering into a clinical translational stage. Both adeno-associated virus (AAV) vectors and lentiviral vector (LV)-mediated hematopoietic stem and progenitor (HSPC) gene therapy have the potential to provide effective therapy for this multisystemic disorder. Optimization of viral vector designs, providing tissue-specific expression and GAA protein modifications to enhance secretion and uptake has resulted in improved preclinical efficacy and safety data. In this review, we highlight gene therapy developments, in particular, AAV and LV HSPC-mediated gene therapy technologies, to potentially address all components of the neuromuscular associated Pompe disease pathology.

|
Болезнь Помпе - редкое аутосомно-рецессивное метаболическое заболевание, вызванное мутациями в ферменте, кодирующем белок кислой альфа-глюкозидазы (GAA) [1]. Фермент GAA активен в кислой среде лизосом и расщепляет гликоген до глюкозы в клеточных лизосомах. Мутация в этом ферменте вызывает накопление гликогена, что приводит к широкому спектру клинических проявлений, определяемых степенью дисфункции белка, наблюдаемой в скелетных, дыхательных и сердечных мышцах [2]. Среди патофизиологических изменений нейровоспаление является отличительной чертой поражения центральной нервной системы (ЦНС), приводящего к нейродегенерации и когнитивным нарушениям [3]. Помимо лизосомной дисфункции, накопление гликогена также приводит к аутофагическому накоплению в мышечных волокнах, что впоследствии может привести к резистентности к методам лечения [4]. Болезнь Помпе в целом классифицируется на две основные категории: болезнь Помпе с младенческим началом (IOPD) и болезнь Помпе с поздним началом (LOPD), которая в значительной степени зависит от остаточной активности фермента GAA. IOPD диагностируется при рождении и при отсутствии лечения приводит к смерти от сердечно-дыхательной недостаточности в течение первого года жизни [2]. При LOPD мышечная деградация развивается медленнее и приводит к нарушению двигательных функций и, в конечном итоге, к дыхательной недостаточности [5,6]. Внутривенная (IV) заместительная ферментная терапия (ERT) является стандартом лечения, направленным на восстановление внутриклеточной активности фермента GAA и устранение клеточной патологии [7]. Тем не менее, ERT имеет риск иммунологических реакций, плохо усваивается в тканях-мишенях и не достигает ЦНС из-за гематоэнцефалического барьера (BBB). Новые разработки в этой области в основном включают адено-ассоциированную вирусную (AAV) генотерапию и лентивирусную (LV) генотерапию в контексте трансплантации (HSCT) гемопоэтических стволовых клеток и клеток предшественников
(HSPC).
Table 1. Overview of academic and company-sponsored preclinical, clinical stage and marketed therapies to treat Pompe disease. Gene therapy applications are in the transitional development stage to clinical application. Clinicaltrial.gov numbers are provided in bold. Ab = antibody, AAV = adeno-associated viral, CMV = cytomegalovirus enhancer/promoter, HSPC = hematopoietic stem and progenitor cell, LV = lentiviral vector, ERT = enzyme replacement therapy, IOPD = infantile-onset Pompe disease, LOPD = late-onset Pompe disease. Yellow = ERT product; Gray = chaperone therapy; Orange = AAV Gene Therapy; Blue = HSPC LV Gene Therapy.
2. Current Standard of Care for Pompe Disease
Enzyme Replacement Therapy
В настоящее время стандартом лечения болезни Помпе является ERT, которая требует пожизненного внутривенного введения рекомбинантного человеческого GAA (rhGAA). В настоящее время альглюкозидаза альфа (Myozyme® или Lumizyme®) является единственным утвержденным препаратом rhGAA для лечения как IOPD так и LOPD. Режим дозирования 20 мг/кг раз в две недели является относительно высоким по сравнению с другими препаратами для лечения lysosomal storage disease (LSD) (табл. 2), и хотя этот режим дозирования обеспечивает эффективное удаление (клиренс) гликогена в сердце и устранение кардиомиопатии [8], он менее эффективен для устранения гликогена в скелетных мышцах и не устраняет респираторное или неврологическое нарушения [9].
Таблица 2. Утвержденные дозы LSDs. Информация взята с сайта accessdata.fda.gov. Burrow и Grabowski сообщили о соотношении U/kg к mg/kg * [10], MPS = мукополисахаридоз. Одной из основных проблем обеспечения длительного бессимптомного выживания при использовании только ERT является ограниченная эффективность доставки rhGAA в пораженные ткани [11], что обусловлено сочетанием факторов, включая низкую концентрацию rhGAA в интерстициальном пространстве, небольшой процент rhGAA, содержащих необходимые модификации манноза-6-фосфата (M6P), необходимые для связывания катион-независимого рецептора манноза-6-фосфата (CI-M6PR), низкое содержание CI-M6PR в клетках скелетных мышц, аномальное перемещение M6P при лизосомных заболеваниях [12] и преимущественное поглощение rhGAA в печени, что приводит к непродуктивному удалению фермента.
Созревание белка GAA происходит через форму предшественника с молекулярной массой 110 кДа [13], который после внутриклеточного протеолитического созревания и гликозилирования в сети Гольджи приводит к появлению активной зрелой форме 70-76 кДа, находящейся в лизосоме [14]. У здоровых людей лизосомная GAA расщепляет гликоген до глюкозы. Однако для эффективного лизосомного нацеливания rhGAA требуется связывание молекул M6P с CI-M6PR на поверхности клетки [15,16]. CI-M6PR, также известный как рецептор инсулиноподобного фактора роста (IGF2R), представляет собой полифункциональный трансмембранный белок с высоким сродством к M6P [15].
В связи с ограничениями, присущими доставке rhGAA в пораженные ткани, были разработаны модификации белка для улучшения клеточного поглощения и биодоступности rhGAA. Два подхода к увеличению связывания rhGAA с CI-M6PR включают добавление фосфорилированных олигосахаридных молекул [17,18], что является стратегией, используемой в препарате второго поколения ERT авалглюкозидаза альфа (Nexviazyme®), недавно одобренном для лечения LOPD и проходящем клинические испытания для лечения IOPD [NCT03019406]), и добавление пептидной метки, состоящей из части инсулиноподобного фактора роста 2 (IGF2), для повышения сродства связывания rhGAA с сайтом связывания IGF2 в IGF2R/CI-M6PR (BioMarin Pharmaceutical). Было проведено исследование безопасности, переносимости, фармакокинетики и фармакодинамики GILT-меченого rhGAA (NCT01230801) и начато исследование фазы 3 у пациентов с LOPD (NCT01924845). Хотя исследование продемонстрировало улучшение силы дыхательных мышц, функции легких и выносливости при ходьбе у пациентов с LOPD, спонсор прекратил программу по причинам, не связанным с опасениями за безопасность пациентов или эффективность препарата.
В доклинических исследованиях мыши Помпе, получавшие нео-rhGAA, сконструированный таким образом, чтобы включать повышенное количество остатков M6P, показали значительно большее снижение гликогена в скелетных мышцах по сравнению с мышами, получавшими rhGAA. Эти мыши также показали сопоставимое снижение гликогена в сердце и диафрагме при 4-кратном снижении дозы [17]. Кроме того, у мышей, получавших нео-rhGAA, наблюдалось улучшение двигательной функции в тестах на ротароду и подвешивание на проволоке, хотя это улучшение не наблюдалось у старых мышей Помпе. В рандомизированном двойном слепом исследовании фазы 3 (NCT02782741), в котором гликоинженерная авальглюкозидаза сравнивалась с существующим стандартом лечения - альглюкозидазой, пациенты, получавшие авальглюкозидазу, продемонстрировали клинически значимое улучшение дыхательной функции по сравнению с пациентами, получавшими альглюкозидазу, а также дополнительное улучшение функциональной выносливости и мышечной силы [19].
Еще один инновационный подход к увеличению биодоступности и улучшению фармакокинетики и фармакодинамики rhGAA - использование небольших молекул шаперонов для стабилизации и усиления ферментативной активности GAA. Шапероны достигают этого путем содействия правильному складыванию белка GAA, что позволяет ему сохранять свою каталитическую активность и предотвращать преждевременную деградацию в эндоплазматическом ретикулуме. Эти шапероны могут вводиться в тандеме с обычной ERT [20] или в контексте нового rhGAA с более высоким уровнем M6P [21]. В дополнение к коррекции накопления гликогена в скелетных мышцах в доклинической мышиной модели Помпе, комбинация нового rhGAA (ATB200), вводимого совместно с шапероном AT2221 (miglustat, N-бутил-дезоксиноджиримицин [NB-DNJ]) смогла уменьшить накопление аутофагии в скелетных мышцах и улучшить wire hang and grip test function (повисания на проволоке и хватания) в доклинической модели мыши, однако результаты клинических испытаний не показали превосходства над существующим стандартом лечения и указывают на необходимость дальнейших долгосрочных исследований (NCT03729362) [22].
Другие новые подходы по доставке rhGAA были оценены на предмет безопасности и эффективности, включая VAL-1221 (Valerion Therapeutics), который представляет собой слитый белок моноклонального волчаночного анти-ДНК-антитела 3E10 с rhGAA, что позволяет модифицированному rhGAA проникать в клетку через equilibrative nucleoside transporter 2 (ENT2), независимо от CI-M6PR. ENT2 экспрессируется на высоком уровне в скелетных мышцах и сердце [23]. Однако это исследование было прекращено спонсором в 2020 году из-за отсутствия финансирования.
rhGAA неэффективно пересекает ВВВ. Для достижения этой цели были изучены стратегии, использующие рецептор-опосредованный эндоцитоз и пути трансцитоза через рецепторы, эндогенно экспрессируемые на эндотелии капилляров мозга [24]. Нацеливание на рецептор трансферрина (TfR) путем слияния rhGAA с античеловеческим антителом к рецептору трансферрина обеспечивает трансцитоз через BBB и доставку в ЦНС [25]. Помимо ограниченной способности современных схем ERT проникать в ЦНС, еще одним ограничением ERT является риск иммунологического ответа. Более высокая доза ERT была благоприятна для клиренса гликогена из скелетных мышц, но приводила к образованию высоких титров анти-rhGAA IgG антител в cross-reacting immunological material (CRIM) отрицательных пациентов Помпе [26]. Такой слабый ответ у CRIM-негативных пациентов Помпе, вероятно, связан с полным отсутствием белка GAA, поэтому у них не развивалась иммунная толерантность. У мышей, лишенных экспрессии GAA, также развивается тяжелый иммунный ответ на введение rhGAA [27].
Продвижение ERT второго поколения, вероятно, повысит эффективность у пациентов с Помпе; тем не менее, сохраняются присущие проблемы и ограничения, такие как повторные вливания в течение всей жизни, иммунологические реакции и неспособность rhGAA пересекать BBB у пациентов с Помпе и патологией ЦНС. Тем не менее, некоторые модификации белка GAA, улучшающие секрецию и клеточное поглощение, могут быть включены в отдельные методы лечения, такие как генотерапия, которая обеспечит постоянный уровень экспрессии выше критического порога, необходимого для предотвращения или коррекции патологии мышц и ЦНС напрямую или посредством перекрестной коррекции (Рисунок 1).

Figure 1. Anticipated pharmacokinetics of enzyme replacement therapy (ERT) using recombinant human GAA (rhGAA) protein and single intervention gene therapy. Left: ERT requires intermittent bolus infusions of doses of rhGAA protein to reach above the critical threshold. It has been reported that above 30% GAA enzyme activity present in unaffected individuals is the critical threshold [1]. Peak plasma rhGAA protein levels are present directly after infusion, subsequently taken up by muscles, and degraded over time. Right: Gene therapy applied as a single intervention therapy for curative potential. After transduction of cells of interest, continuous production of therapeutic transgene product provides sustained levels in transduced cells and/or secreted levels in plasma for cross-correction in key tissues. Application of gene therapy may impact the bioavailability of therapeutic enzymes to enhance uptake and correction in key tissues compared to ERT as shown by Costa-Verdera and colleagues [28]. Horizontal dotted line represents the critical threshold to prevent Pompe disease phenotype. 3. Gene Therapy for Pompe Disease
Доминирующими системами переноса генов, исследованными в доклинических и клинических условиях, являются векторы на основе вирусов, предназначенные для использования в генотерапии in vivo и ex vivo, которая в случае болезни Помпе в основном сосредоточена вокруг адено-ассоциированных вирусных (AAV) векторов для применения in vivo и лентивирусных (LV) векторов для генотерапии ex vivo гемопоэтических стволовых клеток. Обзор современных методов генной терапии болезни Помпе представлен на рисунке 2.
 Figure 2. Overview schema of Pompe disease gene therapy modalities. Left panel represents the process of autologous ex vivo lentiviral gene therapy. Briefly, CD34+ cells are isolated in a closed manufacturing system from mobilized peripheral blood. These isolated cells are transduced with a lentiviral vector containing the functional gene of interest, and the genetically modified cells are infused back into a patient who has typically been conditioned with alkylating agents, such as busulfan, to create space in the bone marrow. Long-term repopulating stem cells engraft into the bone marrow niche and repopulate the hematopoietic system with cells capable of secreting functional enzyme, leading to uptake and cross-correction of affected peripheral tissues. The conditioning agent, busulfan not only makes space in the bone marrow for CD34+ to permanently engraft, but also enables the microglia in the CNS to be exchanged for gene-modified microglia derived from the infused cells. The right panel depicts in vivo AAV gene therapy approaches. Utilization of different AAV vector capsid proteins and routes of administration are used to target distinct tissue niches. Transduced cells secrete functional enzyme locally, or systemically depending on targeted tissue. ICV = intracerebroventricular, IT = intrathecal, IV = intravenous, IM = intramuscular, IP = intraperitoneal. Figure 2. Overview schema of Pompe disease gene therapy modalities. Left panel represents the process of autologous ex vivo lentiviral gene therapy. Briefly, CD34+ cells are isolated in a closed manufacturing system from mobilized peripheral blood. These isolated cells are transduced with a lentiviral vector containing the functional gene of interest, and the genetically modified cells are infused back into a patient who has typically been conditioned with alkylating agents, such as busulfan, to create space in the bone marrow. Long-term repopulating stem cells engraft into the bone marrow niche and repopulate the hematopoietic system with cells capable of secreting functional enzyme, leading to uptake and cross-correction of affected peripheral tissues. The conditioning agent, busulfan not only makes space in the bone marrow for CD34+ to permanently engraft, but also enables the microglia in the CNS to be exchanged for gene-modified microglia derived from the infused cells. The right panel depicts in vivo AAV gene therapy approaches. Utilization of different AAV vector capsid proteins and routes of administration are used to target distinct tissue niches. Transduced cells secrete functional enzyme locally, or systemically depending on targeted tissue. ICV = intracerebroventricular, IT = intrathecal, IV = intravenous, IM = intramuscular, IP = intraperitoneal. 3.1. AAV Vector-Mediated Gene Therapy in Pompe Disease
Методы генотерапии болезни Помпе в основном тестировались на мышах Gaa-/-, широко используемой мышиной модели болезни Помпе [29,30]. Одним из первых подходов генной терапии для лечения мышей Gaa-/- было использование аденовирусного вектора, содержащего усилитель/промотор CMV, стимулирующий экспрессию кДНК GAA, что привело к системному снижению уровня накопленного гликогена. В этом эксперименте однократное внутривенное введение использовалось для проверки способности печени выделять функциональный фермент-предшественник GAA человека, что приводило к высоким уровням активности GAA в плазме крови с последующей перекрестной коррекцией и снижением уровня гликогена в сердце, скелетных мышцах и гладких мышцах [31]. Другой подход использовал в доклинических исследованиях гибридные аденовирусные-AAV векторы, в которых были объединены свойства аденовирусного (Ad) хелперного вируса и упаковки AAV. Эти векторы Ad-AAV-hGAA продуцировали детектируемый белок GAA после направленного на печень воздействия и были способны очищать гликоген в скелетных мышцах мышей с нокаутом GAA [32,33]. С тех пор основное внимание уделялось использованию генотерапии с помощью AAV для лечения болезни Помпе. Векторы AAV содержат икосаэдрический капсид, и на сегодняшний день охарактеризовано несколько серотипов капсида, каждый из которых обеспечивает определенный тропизм к клеткам печени, мышц и тканей ЦНС. Рецепторы или корецепторы, используемые AAV для проникновения в клетки, специфичны для серотипа капсида и отличаются по количеству в зависимости от типа клеток, что может быть использовано для целенаправленного воздействия на ткани, специфичные для конкретного заболевания [34]. Эти серотипы капсида также могут быть дополнительно модифицированы для нацеливания на новые ткани, представляющие интерес. AAV содержит линейный одноцепочечный геном ДНК размером около 4,7 Кб [35], и при удалении генов rep/cap образуется достаточно места для включения кДНК GAA человека (база данных CCDS: CCDS32760.1) размером 2859 нт (952 аминокислоты), фланкированной инвертированными терминальными повторами (ITR). Эта последовательность кДНК GAA может управляться элементами промотора для обеспечения тканеспецифической экспрессии. Естественная инфекция AAV не вызывает никаких заболеваний у человека, но время заражения определяет иммуногенность [36]. Преимуществом векторов AAV является эффективная трансдукция окончательно дифференцированных клеток, обеспечивающая длительную экспрессию, при этом они остаются преимущественно эписомными, что снижает риск инсерционного мутагенеза. Однако сообщается, что векторы AAV обладают способностью интегрироваться в геном хозяина, причем наиболее распространенным сайтом геномной интеграции вируса дикого типа AAV2 является AAVS1 [37]. Считается, что эта способность утрачивается при модификации AAV в векторы генотерапии, поскольку белки AAV Rep, необходимые для интеграции AAVS1, отсутствуют [38]. Совсем недавно клональная экспансия трансдуцированных клеток наблюдалась у собак с гемофилией А, за которыми следили около 10 лет [39]. В геномной ДНК клонально доминирующих клеток были обнаружены интеграции AAV, что подчеркивает необходимость длительного наблюдения за потенциальной генотоксичностью, связанной с векторами AAV.
3.1.1. Muscle Directed Delivery
Генотерапия, направленная на мышцы, может непосредственно исправить патологию пораженной ткани, связанную с болезнью Помпе, и с этой целью несколько векторов AAV с мышечным тропизмом были протестированы в доклинических и клинических исследованиях. Рекомбинантный AAV-вектор, кодирующий кДНК Gaa мыши и упакованный с рекомбинантным AAV1 (rAAV1) серотипа (AAV1/2), был эффективен в обеспечении более высоких уровней фермента и улучшении уменьшения отложений гликогена при внутримиокардиальном и внутримышечном введении Gaa-/- мышам [40]. Эти результаты продемонстрировали первые попытки устранить кардиомиопатию, наблюдаемую у нокаутных GAA мышей и восстановить сократительную функцию скелетных мышц с помощью векторов rAAV1/2. Кроме того, дыхательная недостаточность была типичной отличительной чертой пациентов с болезнью Помпе [41,42], поэтому были проведены исследования диафрагмальной доставки. Таким образом, для улучшения дыхательных функций была предпринята попытка внутримышечной доставки AAV1 в диафрагму. Этот метод использовал гель на основе глицерина для доставки вектора в диафрагму [43], и четко продемонстрировал тропизм серотипа AAV1 в достижении равномерной экспрессии GAA в диафрагме. Этот вектор AAV1-CMV-GAA, стимулирующий экспрессию GAA промотором CMV, был использован в доклинических исследованиях на мышах Gaa-/- [44], а биораспределение/токсикологические исследования, проведенные на новозеландских кроликах, показали распространение вектора по всей диафрагме после локальных инъекций [45]. Результаты показали значительное улучшение дыхательных функций при введении в раннем возрасте, но эффект снижался с возрастом. Интересно, что они наблюдали ослабление активности эфферентного phrenic (диафрагмального) нерва, что указывает на ретроградную трансдукцию, что требует дальнейших подробностей для изучения клиренса гликогена из периферической нервной системы. Сообщалось, что использование промотора CMV для экспрессии трансгенов в системах вирусных векторов подвержено замалчиванию [46], что может объяснить снижение эффекта в этих исследованиях. Вектор rAAV1/2-CMV-GAA также вводили внутривенно однодневным мышам [47]. Этот подход показал коррекцию мышечной функции и улучшение дыхательной и сердечной функций, но преходящий гуморальный ответ наблюдался через 4-18 недель после лечения. Кроме того, вектор AAV1-CMV-GAA был оценен на безопасность при диафрагмальной доставке генов у пациентов с IOPD [48], (NCT 00976352). Целью данного клинического испытания было достижение более высоких показателей дыхания по сравнению с исходным уровнем, а также ожидалось улучшение нервно-мышечной работы phrenic. До 180-го дня у испытуемых наблюдалось значительное улучшение, но к 365-му дню респираторные параметры начали снижаться. Несмотря на обнадеживающие результаты, для поддержания экспрессии GAA может потребоваться повторная дозировка.
В дополнение к этим исследованиям, векторы AAV, кодирующие человеческий GAA, экспрессируемый промотором мышечной креатинин-киназы, были исследованы на мышах GAA-KO [49]. В этих экспериментах мышам внутримышечно вводили вектор, что приводило к успешному снижению гликогена в скелетных мышцах, однако о коррекции ЦНС не сообщалось. В этом случае наличие нейтрализующих антител к hGAA предотвратило перекрестную коррекцию сердечной мышцы. В этом отчете также указано, что выбор промотора зависит от серотипа AAV, и промотор muscle creatine kinase (MCK) был активен в сердце с серотипом AAV2/7, но не с серотипом AAV2/6. Неврологические проявления все еще оставались препятствием для полной нормализации дыхательной дисфункции, связанной с накоплением гликогена в ЦНС.
В другом доклиническом исследовании на мышах Pompe была показана коррекция сердечной и мышечной патологии с помощью конструкции, содержащей промотор десмина (DES) и кодон-оптимизированную последовательность GAA (rAAV9-DES-hGAA). Этот подход обеспечил улучшение функций сердечных, скелетных и дыхательных нейронов у мышей Pompe но экспрессия у мышей с Gaa была преходящей [50]. Промотор десмина демонстрирует более тканеспецифичную экспрессию в скелетных мышцах, сердечной мышце и двигательных нейронах [51] и может снизить иммуногенность по сравнению с использованием векторов с конститутивно активными промоторами, например, CMV или гибридными промоторами куриного энхансера β-актина для экспрессии трансгенов. В данном исследовании использовали rAAV9-DES-hGAA для системного введения мышам Gaa-/- и сравнили результаты с ERT. Улучшение кардиомиопатии наступило не сразу, а через три месяца после инъекции. Модуляция иммуногенности в ответ на GAA была значительно снижена по сравнению с ERT. В последующем клиническом исследовании у пациентов с LOPD была протестирована внутримышечная доставка AAV9-DES-hGAA в переднюю большеберцовую мышцу (NCT02240407) вместе со стратегией иммуномодуляции для уничтожения В-клеток, чтобы позволить повторное введение того же вектора для повышения потенциала вектора для коррекции патологии заболевания в сердечной и скелетной мышцах [52].
В недавнем исследовании на мышах Gaa-/-, а также на не-человекообразных приматах, системное введение препарата AT845, вектора серотипа AAV8, содержащего мышиный промотор/энхансер MCK, управляющий кодон-оптимизированным человеческим GAA, привело к супрафизиологическим уровням активности фермента, что привело к значительному функциональному улучшению и устранению гликогена в ключевых тканях-мишенях дозозависимым образом. Несмотря на хорошую переносимость в низких дозах, высокие дозы у макак циномольгус привели к иммунным реакциям, связанным с вектором, и воспалению, с нарушениями сердечной деятельности, что потребовало внеплановой эвтаназии двух животных из исследования. Позже был сделан вывод, что иммунные реакции были в значительной степени обусловлены ксеногенным иммунным ответом против GAA, поскольку идентичный вектор, несущий GAA, полученный от макак, не привел к таким же воспалительным и сердечным патологиям [53].
3.1.2. Liver Directed AAV Gene Therapy
Печень является важным органом, который естественным образом производит сывороточные белки, и часто используется в качестве депо для эффективного производства рекомбинантных белков для генотерапии. Она также обладает иммунотолерантными свойствами, помогающими предотвратить иммунные реакции, связанные с продуктом трансгена, если введенный ген специфически экспрессируется в гепатоцитах [54]. Специфический для печени промотор (LSP), полученный из промотора тиреоидного гормон-связывающего глобулина и последовательности усилителя a1 микроглобулина/бикунина, был использован в векторе AAV8-hGAA и введен в низкой дозе мышам Gaa-/- и эффективно вызывал индукцию иммунной толерантности у иммунокомпетентных мышей Gaa-/- [55,56]. Кроме того, этот специфический для печени промотор привел к эффективному снижению гликогена в сердце и диафрагме при оценке через 12 недель после генотерапии. Введение низких доз вектора за 6 недель до заражения мышей rhGAA показало отсутствие развития антител, только при использовании гепатоцит-специфического промотора, и поэтому использование AAV2/8-LSP-hGAA может повысить эффективность ERT у CRIM-негативных пациентов Pompe [57]. Кроме того, эффективность конструкции AAV2/8-LSP-hGAA была проверена на средней эффективной дозе на мышах Pompe, и исследователи обнаружили, что доза вектора была обратно пропорциональна выработке анти-GAA антител [58]. Эти доклинические исследования послужили основанием для текущего проспективного испытания с открытой меткой на пациентах с LOPD (NCT 03533673). В другом подходе гибридный тандемный печеночно-мышечный промотор (LiMP) с использованием серотипов AAV8 и AAV9 был использован для неонатальных инъекций мышам Gaa-/- , что обеспечило высокую и стойкую мультисистемную экспрессию трансгена в неделящихся внепеченочных тканях, а также способствовало предотвращению анти-трансгенного иммунитета [59].
Однако, хотя печень эффективно секретирует рекомбинантные белки, белок GAA обычно плохо секретируется из трансдуцированных клеток [60]. Поэтому было исследовано несколько модификаций белка GAA для оптимизации экспрессии трансгена и усиления секреции белка GAA (рис. 3). Sun и др. исследовали, можно ли заменить сигнальный пептид последовательности GAA на сигнальный пептид секретируемого белка [61]. Пять сигнальных пептидов были протестированы в клеточных линиях, и замена сигнального пептида GAA на сигнальный a1 microglobulin/bikunin enhancer (hAAT) привела к усилению секреции белка GAA, а также показала повышенную эффективность у иммунокомпетентных Gaa-/- мышей. В рамках другой стратегии исследовалась доставка rAAV8 с помощью печеночного тропизма, что привело к низкой иммуногенности и частичной перекрестной коррекции патологии ЦНС и мышц [60,62]. В данном исследовании для повышения секреции белка GAA и снижения иммуногенности использовались различные секретируемые сигнальные пептиды и делеция восьми аминокислот в N-концевой части, слитые с кодон-оптимизированным GAA. Серотип AAV8 со специфической для печени аполипопротеиновой контрольной областью и промотором альфа-1-антитрипсина (hAAT), который ранее использовался для направленной на печень генотерапии синдрома Криглера-Наджара [63], показал, что конструкция AAV8-hAAT-sp7-delta8-coGAA была очень эффективна в расщеплении гликогена в мышцах и уменьшении патологии, но уменьшение гликогена в ЦНС было менее заметным [60]. Однако на срезах головного и спинного мозга было обнаружено присутствие лизосомальных GAA и снижение количества Iba1+ клеток, что указывает на влияние на нейропатологию. Высокое количество секретируемого GAA из печени у не-человекообразных приматов и уменьшение количества анти-GAA антител к sp7-delta8-coGAA подтвердили сниженную иммуногенность при повышенной активности GAA в плазме крови и скелетных мышцах. В последующем исследовании секретируемый GAA AAV8 сравнивался с ERT у мышей с ослабленным иммунитетом Gaa-/- Cd4-/- и генотерапией, направленной на печень, и особенно в высокой дозе (2 х 10 12 vg/кг), обеспечивал устойчивую активность GAA в мышцах и снижение гликогена [28]. В ЦНС белок и активность GAA были значительно увеличены в спинном и головном мозге, но эффект на снижение гликогена в головном мозге был менее выражен по сравнению со спинным мозгом.
 Figure 3. Summary of GAA protein modifications employed in ERT and gene therapy. A redrawn and modified model for maturation of GAA protein, previously reported by Moreland et al. [13]. Modifications to GAA transgene or protein described in literature were subject to changing the N-terminus of the GAA protein to improve secretion and uptake in the key tissues affected in Pompe disease without affecting proteolytic processing steps to the mature GAA protein. Signal peptides have been modified to improve secretion, and tags have been incorporated to enhance uptake in skeletal muscles or cellular delivery to cytoplasm to degrade glycogen more effectively. For enzyme replacement therapy enhanced glycosylation or chaperone has been investigated as well. GT = gene therapy, ERT = enzyme replacement therapy, Fab = antigen-binding fragment. scFv = single-chain variable fragment, hAAT = human alpha-1-antitrypsin. В настоящее время компания Spark Therapeutics (NCT04093349) спонсирует исследовательское клиническое испытание с использованием rAAV с биоинженерным капсидом, полученным из Rh74, и кассетой экспрессии для управления экспрессией секретируемого белка GAA (SPK-3006). В недавно опубликованном доклиническом исследовании генотерапии AAV, проведенном Байком и коллегами [64], каталитические части GAA были слиты с одноцепочечным вариабельным фрагментом (scFv), нацеленным на CD63. Антиген CD63 был выбран в результате скрининга, проведенного для отбора эффекторных белков, которые перемещаются с поверхности клетки в лизосому, и которые экспрессируются в скелетных мышцах, но минимально экспрессируются в печени. Этот подход был протестирован в качестве ERT, а также посредством специфической для печени экспрессии AAV8, и показал превосходство в снижении гликогена, уменьшении аутофагического накопления в скелетных мышцах и последующем улучшении функции мышц.
3.1.3. CNS Directed AAV Gene Therapy
Проявления в ЦНС при болезни Помпе очень заметны и являются важным компонентом, который необходимо учитывать при генотерапии пациентов как с IOPD , так и с LOPD [65,66]. Для того чтобы предотвратить, приостановить или потенциально обратить вспять патологию ЦНС, было предпринято несколько попыток с использованием рекомбинантных векторов AAV. Серотипированный AAV5 вектор, кодирующий белок GAA, AAV5-GAA был введен на уровне C3-C4 спинного мозга взрослых мышей с нокаутом GAA, что привело к уменьшению гликогена в шейных вентральных рогах в спинным мозге и впоследствии показало потенциальное улучшение дыхательной функции [67]. Это подчеркивает, что для эффективного лечения болезни Помпе терапия должна быть направлена как на ЦНС, так и на скелетные мышцы. Позже было проведено несколько доклинических исследований с использованием различных серотипов AAV в попытке обойти иммунный ответ и повысить эффективность.
Серотип AAV9 показал эффективный тропизм к ЦНС у новорожденных и взрослых мышей [68], а также к печени и скелетным мышцам [69]. Нейрональный тропизм AAV9 был использован для генотерапии спинальной мышечной дистрофии, экспрессирующей ген survival motor neuron (SMN) с помощью CMV enhancer и куриного β-актин гибридного (CAG) промотора (NCT02122952) [70,71], что привело к недавнему одобрению onasemnogene abeparvovec (Zolgensma). Используя серотип AAV9, Hordeaux и др. [72] применили однократную интратекальную доставку AAV9-CAG-hGAA мышам с Gaa-/- и продемонстрировали неврологическую коррекцию и сопутствующее улучшение сердечных функций.
Накопление гликогена при болезни Помпе происходит в двигательных нейронах, что способствует нервно-мышечной дисфункции [5,60]. Поэтому была предпринята попытка доставки нейрон-специфического промоторного гена синапсина I с помощью конструкции yfAAV9/3-Syn-I-hGAA [73]. В этом исследовании неонатальным мышам Gaa-/- вводили в желудочки головного мозга, что привело к эффективной экспрессии GAA в клетках нейронов, преимущественно в коре и мозжечке. Отложения гликогена не были устранены в печени и четырехглавой мышце, но в головном и спинном мозге они значительно уменьшились, а также уменьшился астроглиоз. Наблюдалось незначительное улучшение мышечной силы, но локомоторная функция, оцениваемая по ротароду, была нормализована. В другом подходе исследовали AAV-B1, серотип, который был выделен из нового капсида AAV, тропного для ЦНС, после одного раунда отбора in vivo из библиотеки капсидов AAV [74]. AAV-B1 эффективно трансдуцировал клетки ЦНС и мышцы, после чего был протестирован на трехмесячных мышах Gaa-/- с использованием промотора десмина для стимулирования экспрессии GAA для коррекции мышц и ЦНС [75]. Этот вектор эффективно трансдуцировал язык, а также показал улучшение патологии дыхательных путей и увеличение силы хвата, в то время как трансдукция сердца очистила гликоген в миокарде, а умеренная трансдукция наблюдалась в двигательных нейронах. Результаты использования нового серотипа AAV-B1 были аналогичны контрольному вектору AAV9.
В другом исследовании, при использовании интралингвального способа введения рекомбинантных векторов AAV1 и AAV9 мышам Gaa-/-, была получена эффективная трансдукция волокон языка, но AAV9 был более эффективен в трансдукции моторных нейронов [76]. Для повышения мышечной эффективности терапевтического белка был разработан GILT-тегированный GAA с частью IGF2, слитой с GAA. Этот GILT-тегированный GAA ранее исследовался для ERT у мышей Помпе и продемонстрировал пятикратную эффективность по сравнению с эквивалентными дозами rhGAA при очистке гликогена в скелетных мышцах [77], а также улучшил дыхательную функцию в модели мышей Помпе [78]. В других моделях LSD, например, у мышей с MPS VII , использование белков, меченных IGF2, для ERT также позволило более эффективно снизить количество продуктов хранения в ключевых тканях [77,79]. Рекомбинантный GILT-меченый GAA также был исследован в клиническом испытании фазы 1/2 (NCT01230801) [80], в котором пациенты продемонстрировали первоначальное улучшение дыхательных показателей, а также ограниченное увеличение выносливости при ходьбе. Однако GILT-меченый GAA (reveglucosidase alfa) вызвал преходящую легкую гипогликемию у нескольких пациентов из-за молекулы IGF2, которая может связываться с рецептором инсулина с низким сродством. Гипогликемия особенно наблюдалась в группах с высокой дозой, получавших 20 мг/кг. Этот IGF2-меченый GAA был включен в вектор AAV9-DES-IGFIIcoGAA, и после интраназального введения IGF2-меченого GAA белок присутствовал в моторных нейронах XII, о чем свидетельствовало иммуногистохимическое окрашивание GAA, повышение активности GAA в лизатах языка и значительное снижение накопления гликогена [81]. Еще один оптимизированный вектор генотерапии, разработанный Amicus Therapeutics/Университетом Пенсильвании с использованием пантропного капсида AAV, несущего инженерный трансген GAA, модифицированный для улучшения секреции и поглощения, был представлен, который эффективно корректировал патологию сердца, ЦНС и мышц после внутривенного введения 2,5 х 1013 геномов вектора/кг молодым мышам Gaa [82-84]. Наконец, новый AAV-капсид из платформы AIM™ AAV векторов нового поколения компании Abeona Therapeutics, как сообщается, имеет улучшенное биораспределение в сердце, мышцах и ЦНС, и в настоящее время находится в доклинической разработке для лечения болезни Pompe [85].
Несколько модификаций серотипов AAV были использованы в испытаниях генотерапии с повышенной безопасностью и долгосрочной эффективностью в органах-мишенях для коррекции патофизиологии в ЦНС, сердце и скелетных мышцах. Также было проведено несколько успешных доклинических попыток с использованием рекомбинантных векторов AAV, секретирующих большое количество GAA и способных напрямую корректировать ЦНС, устраняя дыхательную дисфункцию у мышей с болезнью Pompe. Хотя клинические испытания, оценивающие безопасность, показывают многообещающие результаты, способность вызывать иммунный ответ на капсидный белок и продукт трансгена, а также уже существующие нейтрализующие антитела против AAV могут поставить под угрозу безопасность и клиническую эффективность. Введение векторов AAV могут вызывать воспалительные токсические реакции, которые усиливаются с увеличением дозы вектора, активацию комплемента, цитопению и выраженную гепатотоксичность [86]. Кроме того, системное введение высоких доз 2 х 1014 копий генома на килограмм веса тела варианта AAV9 не-человекообразным приматам и поросятам вызвало токсичность печени и сенсорных нейронов, не зависящую от иммунного ответа на капсид или продукт трансгена [87]. Более того, у пациентов с Х-сцепленной миотубулярной миопатией исследуемый терапевтический препарат MTM1 (миотубулярин) под названием AT132 с использованием AAV8 привел к четырем смертям с дисфункцией печени через 3-4 недели после приема AT132. У всех пациентов были признаки предшествующего внутрипеченочного холестаза, что позволяет предположить, что фон заболевания мог сыграть свою роль, но требуется дополнительное исследование [88].
Предварительное воздействие AAV дикого типа (WT) при естественной инфекции развивает у людей гуморальный иммунитет против капсидного белка [89]. Нейтрализующие антитела против AAV и перекрестно реагирующие антитела различных серотипов AAV блокируют трансдукцию клеток-мишеней и тем самым значительно изменяют терапевтическую эффективность введенных rAAV с трансгеном [90,91]. Капсиды rAAV и продукт трансгена способны активировать как гуморальные, так и клеточные реакции, способствуя активации цитотоксических Т-лимфоцитов (CTL) и белков комплемента [92]. Эти врожденные проблемы, связанные с rAAV, должны быть тщательно изучены, чтобы улучшить общие результаты. Необходимо продолжить анализ ключевой роли активации TLR9 [93] как медиатора клеточного и гуморального иммунного ответа, а также CpG-мотивов в геноме вектора, которые могут стимулировать иммунные ответы, связанные с активацией CD8+ Т-клеток [94]. Внедрение новых технологий, таких как включение в геном вектора коротких олигонуклеотидов ДНК, которые противодействуют активации TLR9, может быть полезным для снижения TLR9-опосредованного иммунного ответа и может позволить вводить пациентам более высокие дозы [95].
3.2. Lentiviral Mediated HSPC Gene Therapy in Lysosomal Storage Disorders
Гамма-ретровирусные и лентивирусные векторы были использованы в генотерапии гемопоэтических стволовых клеток для лечения заболеваний крови и lysosomal storage disorders (LSDs). В испытаниях генной терапии HSPC при Х-сцепленном тяжелом комбинированном иммунодефиците и синдроме Вискотта-Олдрича с использованием гамма-ретровирусных векторов ранней конструкции, которые привели к развитию Т-острого лимфобластного лейкоза у значительной части пациентов [96]. В клиническом испытании синдрома Вискотта-Олдрича у девяти из 10 пациентов, прошедших лечение, наблюдалось успешное приживление, но у семи пациентов развился острый лейкоз, а анализ сайтов интеграции выявил инсерции вблизи протоонкогенов LMO2, MDS1 или MN1 [97]. Однако при ADA-SCID ретровирусная генотерапия не выявила признаков лейкемической трансформации, несмотря на интеграцию в LMO2 и другие протоонкогены [98-101], это указывает на то, что инсерционный онкогенез зависит от вектора и заболевания. В свете этих первых испытаний, с тех пор в этой области отказались от гамма-ретровирусных векторов и вместо них стали использовать более безопасные самоинактивирующиеся векторы третьего поколения self-inactivating (SIN) LV в качестве платформы для HSPC генотерапии.
LV-векторы способны эффективно трансдуцировать как делящиеся, так и неделящиеся клетки, например, нейроны [102], но для эффективной трансдукции ex vivo HSPC требуется дополнительный этап стимуляции цитокинами [103]. Современные векторы LV представляют собой SIN-векторы третьего поколения, в которых упаковочные последовательности и вектор переноса разделены между четырьмя плазмидами для дальнейшего снижения риска рекомбинации [104-106]. SIN-конфигурация long-terminal repeat (LTR) повышает безопасность и позволяет контролировать экспрессию трансгена с помощью внутреннего промотора, а добавление пост-транскрипционного регуляторного элемента вируса гепатита Woodchuck значительно улучшает экспрессию трансгена [107]. Наиболее часто используемым гликопротеином оболочки для HSPC LV трансдукции человека является g-белок вируса везикулярного стоматита (VSVg), который придает широкий клеточный тропизм [108]. Псевдо-типирование векторов LV с VSVg также обеспечивает стабильность векторных частиц, которые можно легко концентрировать до высоких титров с помощью ультрацентрифугирования [109]. Хотя VSVg обычно используется для псевдо-типирования векторов LV [108], другие гликопротеины, такие как RD114/TR и оболочки ретровирусов бабуина, способны эффективно опосредовать трансдукцию человеческих HSPC и могут быть подходящей, даже выгодной альтернативой псевдо-типированию VSVg. Векторы третьего поколения SIN LV в настоящее время являются преобладающей векторной системой, используемой в испытаниях генотерапии HSPC, и демонстрируют повышенную эффективность и сниженный профиль генотоксичности в доклинических и клинических условиях [110].
LV HSPC генотерапия была успешно применена в клинических испытаниях для лечения метаболических заболеваний, таких как metachromatic leukodystrophy (MLD) [111], болезнь Фабри [112] и болезнь Херлера (Hurler) [113], а также пероксисомального расстройства Х-сцепленной cerebral adrenoleukodystrophy (CALD), с зарегистрированным наблюдением в течение 12 лет после трансплантации [114-116]. В целом, более 350 пациентов прошли лечение лентивирусной генотерапией гемопоэтическими стволовыми клетками при моногенных заболеваниях без каких-либо серьезных побочных явлений, связанных с генотоксичностью [117]. Преимущество использования HSPCs основывается на способности трансдуцированных ex vivo HSPCs длительно приживаться в нише костного мозга и давать начало полному репертуару гемопоэтических клеток (включая микроглию в ЦНС), которые затем действуют как фабрики, обеспечивающие локальную доставку фермента в ткани-мишени заболевания, а также системную доставку через циркуляцию в плазме крови. Индукция иммунной толерантности к рекомбинантным терапевтическим ферментам была продемонстрирована при трансплантации аллогенных гемопоэтических клеток при болезни Hurler's [118] и при генотерапии HSPC Фабри [112], при этом антитела против рекомбинантного белка исчезают в течение нескольких месяцев, что делает генотерапию LV HSPC платформой с очень низким риском иммуногенности.
Дополнительным преимуществом генотерапии LV HSPC является способность модифицированных HSPC дифференцироваться в микроглиальные клетки и приживляться в ЦНС [119]. Модифицированные генотерапией LV микроглиальные клетки могут обеспечить надежную экспрессию белка во всей ЦНС и могут обеспечить значительный терапевтический эффект за счет перекрестной коррекции пораженных болезнью глиальных клеток. Эта характеристика генотерапии HSPC была успешно продемонстрирована в доклинических исследованиях генотерапии в мышиных моделях MLD, в которых высокий уровень приживления в костном мозге после кондиционирования бусульфаном был достигнут с помощью генетически модифицированных HSPC микроглии, действующих как источники продукции фермента арилсульфатазы А в ЦНС, что привело к улучшению неврологического дефицита [120,121]. У пациентов с MLD, получавших аллогенную HCT, не было обнаружено доказательств перекрестной коррекции олигодендроцитов и астроглии; однако донорские макрофаги были способны эффективно переваривать накопленные сульфатиды, что могло играть прямую нейропротекторную роль для резидентных олигодендроцитов, способствуя ремиелинизации [122]. Эти результаты подчеркивают способность генотерапии LV HSPC проникать в нишу ЦНС и оказывать терапевтическое воздействие на LSDs с неврологической патологией.
3.2.1. Lentiviral Vector HSPC Gene Therapy for Pompe Disease Treatment
Генотерапия гемопоэтическими стволовыми клетками ex vivo также прошла доклинические испытания для лечения болезни Pompe, результаты которых показали устойчивую системную экспрессию GAA и способность корректировать неврологический дефицит в мышиных моделях болезни Pompe [123-125]. Трансплантация сингенных генетически модифицированных HSPC имеет то преимущество, что вызывает иммунную толерантность к эндогенно секретируемым белкам и вводимым рекомбинантным белкам, о чем ранее сообщалось в нескольких доклинических моделях [123-127]. Этот подход может обеспечить длительный терапевтический эффект путем однократного введения трансдуцированных клеток и потенциально может быть использован для лечения пациентов как с IOPD, так и с LOPD, при условии достижения достаточно высоких уровней экспрессии GAA, превышающих минимальный порог заболевания. Первое доклиническое исследование, в котором сообщалось о генотерапии HSPC при болезни Помпе, было проведено Douillard-Guilloux et al. [123]. Здесь авторы использовали векторы третьего поколения SIN LV, экспрессирующие GAA человека под управлением вездесущего промотора MND (усилитель вируса миелопролиферативной саркомы, область негативного контроля удалена, заменен сайт связывания праймера D1857 rev) для трансдукции HSPC перед введением облученным Gaa-/- мышам. Результаты показали низкую, но обнаруживаемую активность фермента GAA в периферической крови и клетках костного мозга (~50-80% от уровня дикого типа) через 17 недель после введения, а химеризм, определенный в колониеобразующих клетках, составил приблизительно 13-20%. Несмотря на низкий уровень фермента GAA, значительное снижение гликогена наблюдалось в ткани gastrocnemius, но не в сердце, и результаты были сопоставимы с введением только rhGAA. Важно отметить, что у мышей HPSC, получавших генную терапию Gaa-/-, иммунные ответы IgG не обнаруживаются благодаря индукции иммунной толерантности против продукта трансгена - одно из ключевых преимуществ толерогенной природы трансплантации HSPC [123].
В другом доклиническом исследовании вектор, содержащий сильный промотор strong spleen focus forming virus (SFFV), был использован для стимулирования экспрессии трансгена GAA в гемопоэтических клетках [124]. Супрафизиологические уровни активности фермента GAA были достигнуты через восемь месяцев после введения генетически модифицированных клеток, достигнув ~13-48-кратного повышения в периферической крови и селезенке по сравнению с контролем дикого типа. Химеризм костного мозга составлял примерно ~35% при среднем количестве VCN на диплоидный геном 7,3. Активность фермента GAA сохранялась до 18 месяцев после трансплантации, и наблюдалось значительное снижение отложений гликогена в сердце, диафрагме, легких, печени и селезенке, без снижения гликогена в мозге. Наблюдалось функциональное улучшение опорно-двигательной и дыхательной функции, а также обратное развитие ремоделирования сердца. В целом, эта попытка была успешной в частичном восстановлении функций пораженных органов и тканей, но не достигла биохимической коррекции в ЦНС.
Другой модификацией кассеты экспрессии вектора LV было включение кодон-оптимизированного трансгена GAA (GAAco) для улучшения общей экспрессии [125]. Последующее доклиническое исследование продемонстрировало улучшенное производство белка GAA в кровообращении по сравнению с нативной последовательностью GAA, что позволило очистить отложения гликогена в сердце и скелетных мышцах, улучшить локомоторную функцию и устранить гипертрофию сердца при дозе ~7 VCN/dg. Важно отметить, что человеческий белок GAA был также обнаружен в микроглии и астроцитах у мышей после пересадки. Присутствие генетически модифицированной микроглии по всему мозгу у пересаженных Gaa-/- мышей можно четко продемонстрировать (Рисунок 4), а обнаруженный в астроцитах белок GAA свидетельствует об эффективной перекрестной коррекции [125]. Даже при высоком среднем значении VCN анализ сайтов интеграции показал ожидаемый лентивирусный паттерн, без селекции протоонкогенов и без каких-либо побочных эффектов, связанных с генотоксичностью. Тем не менее, VCN/dg в целом были выше, чем максимальное число копий вектора, рекомендуемое FDA, которое составляет <5 копий на диплоидный геном [128]. Более высокий химеризм генетически модифицированных клеток может позволить улучшить клиренс гликогена при более низком среднем VCN, чем было получено в исследованиях Douillard-Guilloux [123], van Til [124] и Stok et al [125].
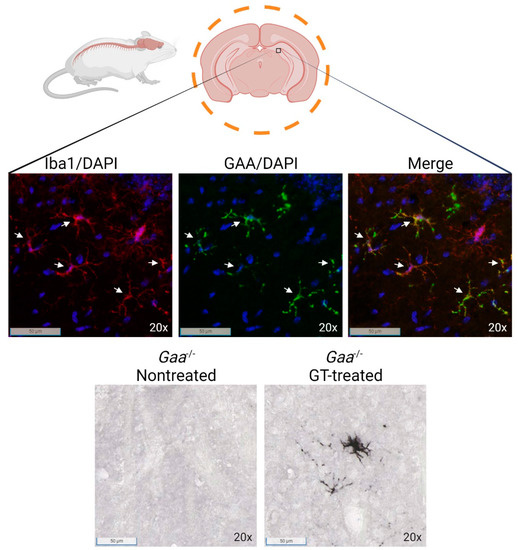
Figure 4. GAA protein expression in resting microglia in the brain of LV HSPC gene-therapy treated Gaa-/- mice. Genetically modified Gaa-/- lineage negative bone marrow cells transduced with a lentiviral vector with codon-optimized GAA, driven by the spleen focus forming virus (SFFV) promoter were infused in to Gaa-/- mice [29] after busulfex conditioning. Brains were harvested at six months after infusion of genetically modified cells in Gaa-/- mice, and post saline perfusion fixed in 4% formaldehyde for 24 h, and subsequently processed for immunofluorescence and immunohistochemical staining for GAA protein. Top panels: Representative anti-GAA (green) and Iba1 (red) immunofluorescence staining shows GAA colocalization in engrafted microglia-like cells in the hippocampal region. DAPI is shown in blue. Bottom panels: representative images of immunohistochemical staining for GAA. White arrows indicate microglia cells colocalizing with GAA signal. Хотя вирусный промотор SFFV был использован в клинических испытаниях для ADA-SCID [129], для клинического применения предпочтительны менее сильные промоторы человека. Поэтому короткий промотор фактора элонгации 1 альфа (EFS) в сочетании с элементами из локуса глобина (LCR-EFS) был использован в генотерапии ADA-SCID для усиления экспрессии в предшественниках эритроцитов [130]. Было высказано предположение, что использование этого слияния усилителя/промотора LCR-EFS может быть полезным и для лечения LSDs. В более поздней публикации вектор LV с эритроид-специфическим элементом LCR-EFS, управляющим кДНК hGAA, частично избавил от клинических проявлений в мышиной модели болезни Помпе [131]. Экспрессия GAA была в 3-6 раз выше в эритроидных клетках, что усиливало секрецию GAA. У мышей с Gaa-/- биохимическая коррекция наблюдалась в сердце, но не в скелетных мышцах, легких и мозге. Соответственно, улучшались параметры сердца, т.е. уменьшение массы сердца после лечения, но без улучшения показателей rotarod или силы хвата. В данном исследовании VCN был относительно низким (~0,6 VCN/dg в крови при диапазоне 0,36-1,44 VCN/dg) при 36-100% химеризме донорских клеток, и может быть выгодно увеличить средний VCN для улучшения биохимической коррекции. Другие конститутивно активные промоторы и/или промоторы с ограничением по линии для обеспечения надежной экспрессии GAA в гемопоэтических линиях могут обеспечить более благоприятный профиль риск/польза. Следует отметить, что человеческие CD34+ HSPC были эффективно трансдуцированы без потери стволовости или потенциала дифференцировки и успешно экспрессировали GAA в плазме крови трансплантированных мышей NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) [131].
Для улучшения доставки в ткани-мишени исследования in vitro показали, что GAAco с меткой IGF2 улучшили секрецию и поглощение в transwell системе [132]. Для этого лентивирусный вектор с промотором MND, стимулирующий экспрессию GAA с меткой IGF2, был использован в генотерапии LV HSPC. В этом исследовании были изучены девять химерных вариантов GAA, управляемых промотором MND [133], включая метки, описанные для улучшения доставки в ЦНС, такие как метка аполипопротеина Е (ApoE) [127] и модифицированная метка IGF2, содержащая мутацию R37A для доставки GILT [134]. Вектор MND-GILT-R37A-GAAco был более эффективен в снижении гликогена в скелетных мышцах и ЦНС, чем вектор GAAco без метки через 16 недель после трансплантации генетически модифицированных HSPC. Эффект в ЦНС был достигнут при относительно низком проценте приживления 0,1-3,7% микроглиальных клеток в мозге, приблизительно рассчитанном с использованием суррогатного контроля Gaa-/- мышей, трансплантированных HSPCs, трансдуцированными вектором GFP.
При использовании трансгена hGAA проникновение в ЦНС и коррекция были минимальными или достижимыми только при очень высоких уровнях системной экспрессии. Предтрансплантационное кондиционирование играет важную роль в успешном приживлении трансдуцированных HSPC в костном мозге и ЦНС [135]. Для того чтобы это было более эффективно, режим кондиционирования должен быть способен уничтожить резидентные микроглиальные клетки и увеличить оборот микроглиальных клеток, полученных от донора, как местных продуцентов терапевтического белка. Режимы кондиционирования, обычно используемые в доклинических моделях животных, включают облучение и химиотерапевтические агенты, такие как алкилирующий агент бусульфан (1,4-бутандиол диметансульфонат). Поскольку микроглия составляет ~5-12% клеток в мозге грызунов [136], терапевтические уровни фермента зависят от процента приживления микроглии в мозге, активности промотора трансгена в микроглиальных клетках и эффективности перекрестной коррекции. Уровень активности фермента GAA в ЦНС мышей Pompe, прошедших лечение, был ниже уровня активности, наблюдаемого у животных дикого типа [133]. Однако низкие уровни активности фермента были аналогичны тем, которые наблюдались в других исследованиях с использованием генотерапии HSPC, например, у мышей Arsa-/- активность фермента арилсульфатазы А достигала почти 10% от активности фермента дикого типа [121], а у обработанных Ids-/- мышей не превышала 4% от уровня дикого типа в мозге. Интересно, что уровни гепарансульфата и дерматансульфата были полностью удалены даже при таких уровнях активности фермента [127].
Исключение из этих уровней фермента наблюдалось у мышей Idua-/-, при генотерапии, что привело к 4,5-кратному уровню от дикого типа [137]. При использовании облучения или кондиционирования бусульфаном количество ген-модифицированных клеток микроглии в мозге было примерно в пределах 10% от клеток микроглии в мозге, что составляет около 1% от общего количества клеток ЦНС. В зависимости от того, какое количество терапевтического белка требуется для достижения эффективной перекрестной коррекции, может быть выгодно увеличить долю генно-модифицированных микроглии в ЦНС.
Колоние-стимулирующий фактор (CSF1R) необходим для выживания микроглии [138], и активность его рецепторной тирозинкиназы может быть подавлена химическим веществом PLX5622, проникающим в мозг. PLX5622 истощает эндогенную микроглию, и его отмена приводит к заселению ниши микроглиальных клеток оставшимися микроглиальными клетками [139], что было продемонстрировано в условиях трансплантации костного мозга мыши, когда обработанные PLX5622 мыши подвергались облучению всего тела в 9 Gy с последующей трансплантацией клеток костного мозга [140]. Через 30 дней ~93% микроглиальных клеток в головном мозге оказались донорскими (~99% в сетчатке и ~93% в спинном мозге). Этот эксперимент показал потенциальное преимущество использования ингибиторов CSF1R для увеличения количества донорских клеток в мозге, что является важным параметром при лечении заболеваний с патологией ЦНС. Необходимы дальнейшие исследования для изучения безопасности этого препарата, но текущие исследования, проверяющие безопасность целенаправленно действующих агентов CSF1R, могут предоставить соединения для увеличения приживления генетически модифицированных клеток в ЦНС [141,142].
Другой подход может заключаться в использовании белков слияния антител к Fc-рецептору новорожденных, белку, связанному с рецептором липопротеина низкой плотности, рецептору трансферрина (TfR) или рецептору инсулина в условиях генотерапии для улучшения доставки непосредственно через BBB в ЦНС, как это было сделано ранее в попытках улучшить ERT [24]. Однако воздействие на рецептор инсулина, как и на другие рецепторы, может быть сопряжено с риском транзиторной гипогликемии и других нежелательных побочных эффектов [143].
3.2.2. In Vivo Lentiviral Gene Therapy for Pompe Disease
В дополнение к подходам лентивирусной генной терапии ex vivo, для лечения болезни Pompe также изучалось прямое применение in vivo. Испытание этого метода in vivo основывается на использовании преимуществ относительно незрелой иммунной системы новорожденных мышей [144]. В соответствии с этой стратегией, Kyosen и др. использовали вектор LV, кодирующий GAA, управляемый промотором CMV, и вводили его непосредственно новорожденным мышам [145]. Обнаруженный в плазме белок GAA стабилизировался с +/- 8 недель, и за ним следили до 24 недель. Результаты показали эффективное снижение гликогена в сердце, но не в диафрагме и четырехглавой мышце. Преимущество неонатального введения заключается в минимизации иммунных реакций, о чем свидетельствует отсутствие инфильтратов CD4+ и CD8+ клеток в тканях, а также малое количество мышей, у которых развиваются анти-GAA антитела. Однако необходимы дальнейшие усовершенствования для эффективного удаления отложений гликогена из тканей сердца, дыхательных путей, скелетных мышц и ЦНС. К сожалению, лентивирусная генотерапия in vivo при болезни Помпе достигла лишь низкой эффективности. Возможно, эта платформа больше подходит для заболеваний, не требующих таких высоких уровней белка, как при болезни Помпе. Лентивирусная генотерапия была исследована в доклинических моделях гемофилии у собак на крупных животных [146], в которых использовались псевдо-типированные лентивирусные векторы VSVg с усиленным гепатоцит-специфическим промотором транстиретина, управляющим оптимизированным по кодонам гиперфункциональным фактором IX собаки, несущим мутацию R338L, и четырьмя тандемными повторами целевых последовательностей miR-142 для ограничения экспрессии в антиген-презентирующих клетках. После внутрипеченочного введения в воротную вену этот подход позволил достичь 1% нормальной активности FIX, однако производственные мощности и наблюдаемые осложнения, связанные с инфузией, могут ограничить применение лентивирусной генотерапии в контексте болезни Помпе. Более поздние модификации клеток-производителей лентивирусных векторов с целью изменения состава лентивирусных частиц путем удаления антигенов лейкоцитов человека и включения сигналов "не ешь меня" для предотвращения фагоцитоза клетками Купфера значительно улучшили трансдукцию в печень [147]. К сожалению, такой подход, направленный на печень, не смог бы эффективно воздействовать на отдел ЦНС, что было бы необходимо для решения проблемы патологии ЦНС, связанной с болезнью Помпе. Подобно этому затруднению, при нацеливании на отдел ЦНС, как было показано на модели Краббе для нечеловеческих приматов, которая продемонстрировала эффективную трансдукцию лентивирусного вектора на нейроны, астроциты и олигодендроциты вблизи места инъекции, не удается нацелить на ключевые ткани на периферии [148].
3.3. Alternative Applications to Modulate GAA Mutations and Disease Correction
В дополнение к подходам клеточной и генной терапии, для лечения PD изучаются альтернативные стратегии на основе молекул. Например, химически модифицированные antisense oligonucleotides (AON), нечувствительные к РНК-опосредованной деградации, были протестированы в доклинических условиях для повышения эндогенной продукции ферментов GAA дикого типа [149,150]. Van der Wal и др. выявили перекрывающиеся AONs, которые повышали активность GAA в клетках, полученных от пациентов, несущих широко распространенный вариант сплайсинга выше пороговых значений заболевания, путем блокирования негативного элемента сплайсинга, что привело к включению экзона 2. Эти результаты являются доказательством концепции использования этого экспериментального метода для коррекции аберрантного сплайсинга и опосредованного включения экзона. Важно отметить, что лечение AONs хорошо переносится в клинических испытаниях при спинальной мышечной атрофии и мышечной дистрофии Дюшена [151,152]. Однако, поскольку AONs специфичны для конкретной последовательности, они обычно подходят только для небольшой подгруппы пациентов с аналогичными вариантами генов, что ограничивает возможность их использования для более широкой популяции пациентов. Этот терапевтический подход также не сможет обеспечить супрафизиологическую экспрессию, необходимую для предотвращения или обратного развития наиболее тяжелых форм PD. Однако альтернативные методы терапии в сочетании с более мощными формами терапии потенциально могут принести пользу пациентам с PD.
Нацеливание на активацию сателлитных клеток при болезни Помпе - еще одна активная область исследований, которая потенциально может быть использована в сочетании с вышеупомянутыми генотерапиями. Скелетные мышцы могут восстанавливаться в ответ на повреждение путем привлечения активности тканевых стволовых клеток. Стволовые клетки скелетных мышц (MuSCs), также известные как клетки-сателлиты мышц, быстро активируются после обнаружения повреждения ткани и размножаются для регенерации поврежденных миофибрилл. Хотя было показано, что количество МСК в биоптатах пациентов с PD остается стабильным, Schaaf и др. обнаружили, что у МСК отсутствуют маркеры активной регенерации, а именно экспрессия эмбриональной тяжелой цепи миозина [153]. Аналогичным образом, в мышиных моделях Помпе мыши с дефицитом Gaa также демонстрируют истощение мышц и отсутствие активации и регенерации МСК, несмотря на исключение внутриклеточных дефектов [154]. Неспособность эффективно активировать МСК для восстановления регенерации мышц, вероятно, частично способствует продолжающемуся истощению мышц, наблюдаемому у пациентов с Помпе, и предполагает, что окружающая ниша может подавлять активацию МСК. Эти данные дают терапевтическое обоснование для изучения методов воздействия на МСК в надежде ослабить мышечную патологию при PD. Было показано, что сателлитные клетки безопасно и эффективно активируются с помощью физических упражнений [155,156]. Предыдущие программы физических упражнений оказались полезными для взрослых пациентов с Помпе и представляют собой разумную дополнительную терапию. Еще одной возможностью является применение малых молекул, нацеленных на активацию MuSCs или косвенно воздействующих на них через связанные пути, такие как аутофагический поток.
Использование стратегий белковой инженерии также показало перспективность в создании более стабильных белков GAA, что дополнит усилия как в области ERT, так и генотерапии. Используя методы направленной эволюции и высокопроизводительного скрининга, Dellas и др. и Botham и др. продемонстрировали, что модифицированные аминокислотные последовательности GAA, содержащие до 30 аминокислотных изменений по сравнению с эталонной последовательностью, могут значительно повысить стабильность белка в нейтральном и низком диапазонах pH, увеличить экспрессию и клеточное поглощение в фибробластах и миобластах при болезни Помпе [157,158]. Эти результаты необходимо подтвердить в естественных условиях, чтобы доказать значимую эффективность и безопасность замены значительного количества аминокислот; однако эта стратегия подчеркивает еще одно интересное направление, в котором инновации могут помочь использовать существующие методы лечения.
Последние достижения в области методов редактирования генов открывают новые захватывающие возможности для лечения генетических нарушений обмена веществ. Хотя in vivo нацеливание на неделящиеся ткани остается сложной задачей, новые подходы к редактированию оснований предлагают решения для подгруппы заболеваний, при которых гомологично-направленная репарация и двуцепочечные разрывы ДНК не нужны. Редактирование оснований позволяет редактировать геном независимо от HDR и разрывов double-stranded DNA с помощью цитидин- или аденозиндезаминазы, слитой с каталитически неактивным Cas9 [159]. Villiger и др. недавно продемонстрировали осуществимость этого подхода, скорректировав фенилкетонурию в мышиной модели аутосомно-рецессивного заболевания печени человека с помощью CRISPR/Cas-ассоциированных редакторов оснований [160]. Используя новый редактор оснований с intein-split , Villiger и коллеги смогли обойти ограничения на размер груза AAV и доставить слитый белок в двух частях. Доставка системы AAV-редакторов оснований привела к физиологическому уровню фенилаланина в крови ниже 120 мкмоль/л, восстановлению активности фермента и реверсии фенотипа светлого меха.
Теоретически, редактирование генома при болезни Помпе было бы привлекательной стратегией, поскольку это позволило бы навсегда исправить мутации в гене GAA и восстановить функцию фермента под нормальным физиологическим контролем. Однако, учитывая, что для того, чтобы остановить прогрессирование заболевания и обратить патологию вспять, потребуется широкое биораспространение и чрезвычайно высокая эффективность коррекции как в мышечной ткани, так и в ткани ЦНС, применение этой технологии, особенно с использованием гомологически-направленной репарации, для коррекции мутаций при болезни Помпе в настоящее время не представляется возможным. Хотя методы редактирования оснований показали эффективность при других заболеваниях, как упоминалось выше, использование редактирования оснований для лечения PD будет затруднено из-за широкого спектра мутаций, наблюдаемых в гене GAA в популяции пациентов, для каждой из которых потребуется уникальная последовательность направляющей РНК. Однако использование стратегий редактирования генома на основе CRISPR может оказаться полезным в условиях генотерапии ex vivo, поскольку это снимет любые опасения по поводу безопасности, связанные с интеграцией генотоксичности, обусловленной доставкой генов с помощью ретровирусных векторов, хотя нежелательные внецелевые мутации все еще должны быть исключены.
Наконец, доступность инструментов CRISPR для создания новых мышиных моделей, которые лучше повторяют фенотипы заболеваний человека, может помочь ускорить разработку следующего поколения терапий PD. Huang и др. недавно сообщили о создании новой мышиной модели IOPD с использованием двойной sgRNA CRISPR-Cas9 гомологически-направленной рекомбинации для укрытия ортологичной мутации Gaa c.1826dupA (p.Y609 *) [161]. Эта модель эффективно повторяет специфический для пациента генотип, который приводит к гипертрофической кардиомиопатии и слабости скелетных мышц - отличительным признакам IOPD человека. Эта модель мыши может быть полезной для тестирования стратегий редактирования генов и редактирования оснований и может быть использована для поиска более эффективных методов лечения.
4. Conclusions
ERT является текущим стандартом лечения IOPD и LOPD, но, к сожалению, несмотря на то, что это большой прогресс в лечении пациентов с Помпе, ERT все еще имеет значительные недостатки, включая способность воздействовать на ЦНС. Появление генотерапии открывает большие перспективы для потенциальной профилактики, остановки и/или обращения вспять болезни Помпе. По сравнению с другими лизосомальными заболеваниями, болезнь Помпе требует большего количества функционального рекомбинантного человеческого белка GAA, распределенного по всему организму, включая ЦНС. Потенциальным решением является генотерапия. Векторы AAV или опосредованная LV HSPC генотерапия могут обеспечить улучшенную биодоступность трансгенного продукта для повышения эффективности. Несколько факторов для генотерапии in vivo с помощью AAV, таких как количество доз, биораспределение и иммуногенность вектора и трансгенного продукта, должны быть тщательно оценены, но технологический прогресс в дизайне вектора и трансгена может ограничить связанные с вектором риски иммуногенности и повысить эффективность. В качестве альтернативы, генотерапия LV HSPC может обеспечить необходимую системную доставку, с оптимизацией кондиционирования и эффективности трансдукции HSPC для максимального приживления генетически модифицированных клеток на периферии и в ЦНС для достижения оптимальной эффективности. Соотношение пользы и риска, связанное с методами лечения, в которых основную роль может играть фон заболевания, требует тщательного изучения каждого решающего шага доклинической разработки. Генотерапия потенциально может быть однодозовым лечением, обеспечивающим пожизненную терапевтическую экспрессию GAA с возможностью предотвращения, остановки или обратного развития болезни Помпе.
|