Анемия - это состояние, при котором количество красных кровяных телец (RBCs и, следовательно, их способность переносить кислород недостаточны для удовлетворения физиологических потребностей организма. Анемия может возникать из-за снижения или нарушения эритропоэза, повышенного разрушения RBCs или кровопотери (ВОЗ, 2015). Симптомы анемии возникают из-за нарушения доставки кислорода тканям или вследствие перегрузки железом, вызванной массивным разрушением RBCs. Уменьшение количества эритроцитов нарушает транспорт кислорода. Следовательно, это может вызвать слабость, утомляемость, трудности с концентрацией внимания и/или плохую работоспособность. Кроме того, перегрузка железом может вызвать хронические заболевания печени и циррозную сердечную недостаточность среди других производных патологий. Хотя основной причиной анемии во всем мире является дефицит железа, существуют и другие ведущие причины анемии, включая наследственные заболевания, такие как гемолитические анемии, которые возникают в результате повышенного разрушения RBCs (Cazzola, 2020). Среди этих врожденных заболеваний RBCs наибольшую частоту встречаемости имеют гемоглобинопатии. Особенно высока заболеваемость серповидно-клеточной болезнью (SCD), поскольку ежегодно рождается около 300 000 больных SCD1 , в основном в Африке и Индии. Это рецессивное генетическое заболевание, вызванное серповидной мутацией в гене β-глобина - изменением аминокислоты с полярного остатка, глутаминовой кислоты (E), на гидрофобный, валин (V), обычно называемый "E6V". Признаки SCD включают низкое количество эритроцитов в крови (анемия), повторяющиеся инфекции и болезненные вено-окклюзионные эпизоды, возникающие, когда эритроциты застревают в мелких кровеносных сосудах.
Несмотря на большое количество пациентов, страдающих наследственными анемиями, единственный метод лечения тяжелых пациентов, страдающих наследственными заболеваниями RBCs, ограничивается аллогенной трансплантацией гемопоэтических стволовых клеток (HSCT), которая имеет особенно серьезные побочные эффекты у этих пациентов. Однако за последние несколько лет появились новые и интересные варианты терапии этой группы заболеваний, а также других наследственных патологий кроветворения. Внедрение новых диагностических технологий, таких как секвенирование следующего поколения (Kaestner and Bianchi, 2020), а также разработка новых терапевтических подходов, таких как генотерапия (Bueren et al., 2020), значительно расширили знания о наследственных анемиях и проложили путь к их окончательному излечению. В настоящее время эти две области совпали, что позволило определить конкретные генетические изменения, которые вызывают различные наследственные анемии, что дает возможность разработать новые стратегии генотерапии [обзор сделан Ferrari et al. (2021)]. Генотерапия облегчает лечение моногенных заболеваний крови путем аутологичной HSCT генно-корректированных гемопоэтических стволовых клеток и клеток предшественников (HSPCs), когда малое количество HLA-совместимых доноров HSPC и риск развития болезни "трансплантат против хозяина" (GvHD) препятствуют использованию аллогенной HSCT. HSPC пациентов очищаются, затем подвергаются генной коррекции
ex vivo перед повторным введением пациентам. Аутологичные ген-откорректированные HSPC приживаются в костномозговой нише пациента, начинают пролиферировать и дифференцироваться для восстановления кроветворения (рис. 1). Эти исправленные HSPC имеют потенциал для пожизненного поддержания кроветворной системы пациента. Поэтому все новые кроветворные клетки, включая RBCs, получаются из исправленных HSPC и несут генетическую коррекцию для восстановления нормальных функций гена, мутировавшего при наследственном заболевании. Гемоглобинопатии были первыми наследственными заболеваниями красной крови, для лечения которых применялась генотерапия, из-за большого количества больных и глубоких знаний об их генетических причинах.
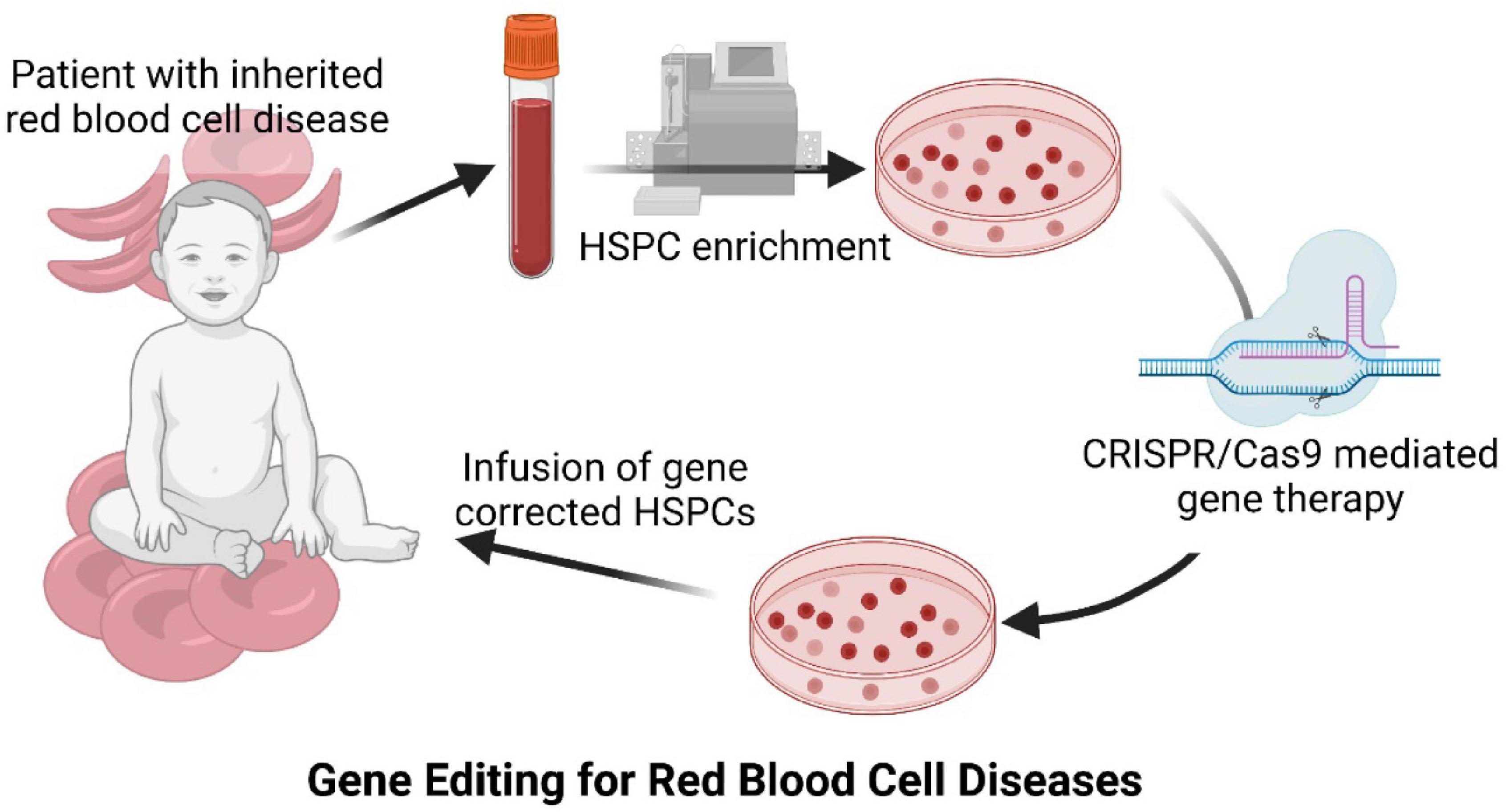
FIGURE 1
Diagram of CRISPR/Cas9 mediated gene therapy approach for inherited red blood cell diseases. С добавлением (с помощью коррекции лентивирусным вектором) и редактирующая гены терапия - две альтернативы для решения проблемы наследственных заболеваний кроветворной системы с помощью генотерапии.
Additional Gene Therapy: Current Successes
Аутологичная HSCT генетически скорректированными клетками станет окончательным методом лечения наследственных анемий, который позволит преодолеть ограничения аллогенной HSCT, такие как ограниченная доступность HLA-совместимых доноров, инфекции, связанные с иммуносупрессией или развитием GvHD. Вирусные векторы семейства Retroviridae были выбраны для доставки терапевтических генетических материалов благодаря их способности интегрироваться в геном клетки-хозяина, что облегчает коррекцию трансдуцированных клеток и всех их потомков (Belmont et al., 1986; Hock and Miller, 1986; Kantoff et al., 1986; Keller and Wagner, 1986; Dzierzak et al., 1988; Hughes et al., 1989; Lim et al., 1989; Szilvassy et al., 1989). Это особенно важно в случае высоко-пролиферативных тканей с небольшим числом стволовых клеток, ответственных за поддержание всей системы, как, например, в кроветворной системе. В таких системах генетическая коррекция популяции стволовых клеток будет передаваться всем различным и функциональным зрелым клеткам. Первые работы показали осуществимость этой стратегии с использованием гамма-ретровирусных векторов (Williams et al., 1984), а первые клинические испытания продемонстрировали их клиническую применимость (Dunbar and Kohn, 1996; Abonour et al., 2000; Cavazzana-Calvo et al., 2000; Aiuti et al., 2002; Hacein-Bey-Abina et al., 2002).
Однако один из лучших клинических успехов генотерапии - излечение пациентов с Х-сцепленным тяжелым комбинированным иммунодефицитом (X-SCID) - оказался серьезным узким местом в этой области, поскольку у некоторых пациентов развилась лейкемия, вызванная гамма-ретровирусным инсерционным мутагенезом (Hacein-Bey-Abina et al., 2003; Howe et al., 2008). Дополнительные фундаментальные исследования новых векторных основ (лентивирусных векторов) и новых регуляторных последовательностей (эндогенных и менее мощных промоторов) помогли вновь продвинуть перманентную генотерапию вперед. В настоящее время лентивирусная генотерапия является предпочтительным вариантом генетической коррекции HSPCs. Более 350 пациентов с различными патологиями были успешно вылечены с помощью этой стратегии. Многие наследственные гематологические заболевания (Bueren et al., 2020; Ferrari et al., 2021), включая гемоглобинопатии, такие как β-талассемия (Cavazzana-calvo et al., 2010; Thompson et al., 2018; Locatelli et al., 2021; Magrin et al., 2022) или SCD (Holmes et al., 2017; Ribeil et al., 2017; Kanter et al., 2021; Magrin et al., 2022), были рассмотрены (Рисунок 1). Фактически, лентивирусами скорректированные HSPC были одобрены для клинического применения у пациентов с трансфузионно-зависимой β-талассемией (Zynteglo2). Талассемия возникает, когда из-за мутаций в гене HBB вырабатывается аномальная форма или недостаточное количество гемоглобина. Мутации HBB вызывают снижение или отсутствие синтеза бета-цепей гемоглобина, что приводит к различным последствиям - от тяжелой анемии до клинически бессимптомного состояния. Ежегодная заболеваемость в мире оценивается как один случай на 100 000. Страдающие этим заболеванием люди также страдают от анемии, которая может вызывать бледность кожи, слабость, усталость и более серьезные осложнения. Поскольку мы сосредоточились на наследственных гемолитических анемиях, недавно мы разработали лентивирусный вектор для генетической коррекции дефицита пируваткиназы (PKD) (Garcia-Gomez et al., 2016). PKD - это аутосомно-рецессивное заболевание, вызванное мутациями в гене пируваткиназы (PKLR), который кодирует эритроидный белок пируваткиназы (RPK). RPK отвечает за поддержание нормального уровня АТФ в эритроцитах. Заболевание становится клинически значимым, когда активность белка снижается ниже 25% от нормальной активности в эритроцитах. Наиболее частыми клиническими признаками являются легкая или очень тяжелая анемия, ретикулоцитоз, спленомегалия и перегрузка железом, которая может быть опасной для жизни у пациентов с тяжелой формой заболевания. PKD считается наиболее распространенной причиной chronic non-spherocytic hemolytic anemia (CNSHA), распространенность которой составляет примерно 1:20 000. В настоящее время проводится клиническое испытание I фазы лентивирусной генотерапии для оценки ее безопасности и эффективности (NCT04105166). Предварительные данные первых двух пациентов с PKD, которым были трансплантированы аутологичные HSPC с лентивирусной коррекцией, показали быстрое гематологическое восстановление после трансплантации (13 дней) и полное восстановление эритроидных параметров более чем через 18 месяцев после трансплантации (Shah et al., 2021), что свидетельствует о возможности применения этой стратегии для лечения гемолитических анемий (рис. 1).
К сожалению, недавно сообщалось о неконтролируемой клеточной пролиферации, приводящей к пред-лейкемическим или лейкемическим событиям, у пациентов, трансплантированных лентивирусными Т-клетками (Fraietta et al., 2018) и HSPCs (Jofra Hernandez et al., 2021). Этот момент должен быть тщательно изучен в случае пациентов, страдающих гемоглобинопатиями, особенно SCD, и получающих лечение лентивирусной генотерапией (Goyal et al., 2021). Несмотря на то, что количество нежелательных явлений очень низкое, все еще существуют важные проблемы для обеспечения наиболее безопасной и эффективной генотерапии. Среди прочих, наиболее актуальными являются исключение инсерционного онкогенеза и эндогенная регуляция терапевтического гена.
Gene Editing Technology
Первые представления о направленной модификации генов, или редактировании генов, возникли в 1960-х и начале 1970-х годов, когда для манипуляций с ДНК использовались ферменты рестрикции (Smith and Wilcox, 1970; Scherer and Davis, 1979), которые создавали специфические разрезы в ДНК. В следующем десятилетии Capecchi продемонстрировал, что новые последовательности ДНК могут быть введены в геном путем гомологичной рекомбинации (HR), которая позволяет переносить любые модификации в геном живой клетки (Capecchi, 1989). В 1985 году Smithies представил направленные геномные изменения в контексте заболевания крови: вставка последовательности плазмидной ДНК в хромосомный локус β-глобина человека для исправления вредных мутаций у пациентов с талассемией и SCD (Smithies et al., 1985). Однако эффективность HR была очень ограничена в эукариотических клетках. Решение этой проблемы появилось в конце 1980-х - начале 1990-х годов, когда было обнаружено, что индукция двунитевого разрыва (DSB) эффективно усиливает HR на конкретной геномной мишени (Rudin et al., 1989; Rouet et al., 1994). DSB запускает эндогенные механизмы репарации ДНК клетки к месту, где был индуцирован разрыв. При этом действуют различные механизмы. Негомологичное соединение концов (NHEJ) и микрогомологичное соединение концов (MMEJ) включают модификацию двух разорванных концов без шаблона, чтобы сделать их совместимыми для воссоединения (Lieber et al., 2003; Guirouilh-Barbat et al., 2004). NHEJ непосредственно соединяет разорванные концы без шаблона. Однако MMEJ восстанавливает разорванные концы путем выравнивания микрогомологических последовательностей, которые образуются в результате резекции концов (San Filippo et al., 2008; Truong et al., 2013; Chang et al., 2017; Pickar-Oliver and Gersbach, 2019). При HR разорванная нить полагается на репарацию, осуществляемую по шаблону (Szostak et al., 1983). Были разработаны различные механизмы на основе эндонуклеаз для внедрения DSB с высокой точностью. Все различные типы эндонуклеаз способны точно модифицировать геномную ДНК, удалять, вставлять или даже заменять фрагменты ДНК с высокой степенью направленности и отбора.
Meganucleases
Мегануклеазы - это ДНК-нуклеазы, обладающие способностью расщеплять dsDNA в специфических сайтах узнавания. Основным преимуществом этой технологии является высокая специфичность к ДНК мишени благодаря большим сайтам узнавания [14-40 пар оснований (bp)]. Мегануклеазы включают такие ферменты, как I-SceI и I-CreI, которые могут быть использованы для нацеливания на гены в клетках млекопитающих (Cohen-Tannoudji et al., 1998; Grizot et al., 2010). Хотя существует множество доступных природных мегануклеаз, вероятность найти фермент, нацеленный на нужный локус, очень мала, что ограничивает их использование (Fenina et al., 2012). Были разработаны процессы молекулярной инженерии, позволяющие конструировать инженерные мегануклеазы. Однако производство специализированных мегануклеаз остается сложным и крайне неэффективным, что ограничивает их широкое применение.
Zinc Finger Nucleases
Нуклеазы с цинковыми пальцами (ZFN) могут быть сконструированы для воздействия на определенные последовательности ДНК. Группа Chandrasegaran's в 1996 году сообщила о создании первой ZFN путем присоединения белковых мотивов цинковых пальцев (которые обеспечивают селективность связывания ДНК) для расщепляющего домена эндонуклеазы FokI (Kim et al., 1996). В 2003 году Porteus and Baltimore продемонстрировали, что ZFN улучшает нацеливание генов в клеточных линиях человека (Porteus and Baltimore, 2003). Первое клиническое испытание с использованием ZFN было посвящено разрушению CCR5 (корецептора ВИЧ) (Tebas et al., 2014). ZFN были использованы для разрушения эритроидного энхансера гена BCL11A, который является репрессором γ-глобинов во взрослых эритроцитах, в HSPCs для увеличения экспрессии фетального глобина и уменьшения sickling в эритроидных клетках в качестве генотерапии для пациентов с SCD (Holmes et al., 2017). В настоящее время этот подход оценивается в двух клинических испытаниях фазы 1/2a для трансфузионно-зависимой β-талассемии (NCT03432364) и SCD (NCT03653247). Однако токсичность ZFN из-за расщепления в нежелательных местах генома (Porteus and Baltimore, 2003) и его сложная конструкция (Bae et al., 2003; Maeder et al., 2008) препятствуют его более широкому использованию.
Transcription Activator-Like Effector Nucleases
Чтобы преодолеть ограничения ZFNs, была разработана новая серия нуклеаз, названных транскрипционными активатор-подобными эффекторными нуклеазами (TALENs) (Christian et al., 2010; Li et al., 2011). Подобно ZFNs, TALENs представляют собой сконструированные ферменты рестрикции, слитые с каталитическими доменами эндонуклеазы FokI и функционирующие как димеры для расщепления целевого участка мишени ДНК. Было показано, что эта система эффективно воздействует на геномы как соматических клеток человека (Miller et al., 2011), так и плюрипотентных стволовых клеток человека (Hockemeyer et al., 2011). Терапевтический потенциал технологии TALEN был также продемонстрирован после публикации клинического испытания редактированных генов химерных антигенных рецепторных Т-клеток (CART-клеток) для лечения В-клеточного острого лимфобластного лейкоза (Qasim et al., 2017). Совсем недавно технология TALEN была также применена для лечения наследственных заболеваний RBCs, таких как гемоглобинопатии (Lux et al., 2019) и PKD (Garate et al., 2015; Quintana-Bustamante et al., 2019).
CRISPR/Cas System
Система CRISPR/Cas [Clustered Regularly-Interspaced Short Palindromic Repeats and Cas (CRISPR associated) nucleases] является самой недавно разработанной платформой в области редактирования генома. Система CRISPR/Cas изменила ландшафт редактирования генов. Эта новинка расширила потенциальные терапевтические возможности применения редактирования генов для коррекции наследственных заболеваний. Открытие CRISPR началось с бактерий в 1993 году (Mojica et al., 1993). Однако только в 2012 году эта система была разработана как инструмент редактирования генома (Jinek et al., 2012). Распознавание ДНК системой CRISPR/Cas основано на взаимодействии РНК-ДНК. Система CRISPR/Cas9 состоит из нуклеазы Cas9 и single-guide RNA (sgRNA). sgRNA - это сконструированная молекула одиночной РНК, содержащая части crispr РНК (crRNA) и tracr РНК. Single-guide RNA распознает последовательность мишень путем стандартного сопряжения оснований Уотсона-Крика. За ней должен следовать мотив ДНК, называемый protospacer adjacent motif (PAM). Эта последовательность расположена непосредственно ниже последовательности-мишени в геномной ДНК, на нецелевой нити. Нуклеаза Cas9 распознает это взаимодействие и затем осуществляет разрезанию ДНК, что приводит к образованию DSB.
Система CRISPR/Cas9 широко используется благодаря своей простоте и универсальности. Системы CRISPR/Cas обладают терапевтическим потенциалом для генетической коррекции наследственных заболеваний RBC, в основном гемоглобинопатий, которые стали рабочей площадкой для опробования новых подходов к редактированию генов. Вкратце, система CRISPR/Cas9 была использована для ре-экспрессии фетальных глобинов либо путем воссоздания наследственной персистенции фетального гемоглобина (HPFH) (Ye et al., 2016), либо путем нокаута репрессора фетального глобина BCL11A (Canver et al., 2015; Brendel et al., 2016; Wu et al., 2019). Эта стратегия редактирования генов является одной из наиболее перспективных терапевтических альтернатив для лечения гемоглобинопатий. Последняя стратегия уже продемонстрировала клиническую пользу в коррекции β-талассемии и SCD у пациентов (Frangoul et al., 2021). Более того, мутировавший ген HBB был подвергнут генному редактированию для восстановления полностью функционального белка β-глобина путем коррекции серповидноклеточной мутации (Dever et al., 2016; DeWitt et al., 2016; Hoban et al., 2016; Lattanzi et al., 2021; Wilkinson et al., 2021). Кроме того, другое наследственное эритроидное заболевание, PKD, было предложено корректировать с помощью редактирования генов CRISPR/Cas (Fananas-Baquero et al., 2021).
Сегодня описаны и адаптированы для редактирования генов новые системы CRISPR/Cas. Появились различные вариации оригинальной системы CRISPR/Cas9 для расширения набора инструментов редактирования генов. CRISPR-ассоциированные транспозазы (Klompe et al., 2019), редакторы оснований (Komor et al., 2016; Gaudelli et al., 2017) и редактирование праймеров (Anzalone et al., 2019), использовали свойство нацеливания системы CRISPR/Cas9 для получения более точной модификации генов (Li et al., 2021; Newby and Liu, 2021).
Delivery of Gene Editing Tools
Идеальный вектор должен доставлять компоненты редактирования генов в определенный тип клеток, вмещать чужеродные гены достаточного размера, обеспечивать уровень и продолжительность экспрессии трансгена, достаточные для исправления дефекта, и быть неиммуногенным. Основным препятствием для рассмотрения редактирования генов в качестве широкого терапевтического варианта является наличие хорошего метода доставки инструментов редактирования генов в целевую клетку и целевую ткань. Компоненты редактирования генов должны быть перенесены в интересующую клетку/ядро с помощью стратегий ex vivo или in vivo. Это узкое место становится еще более важным, когда рассматривается редактирование генов в HSPCs. Примитивные HSPC трудно трансфицировать и они не поддаются трансдукции вирусными векторами. Необходимо учитывать несколько факторов, включая физические барьеры (клеточные мембраны, ядерные мембраны), стабильность инструментов редактирования генов внутри клеток-мишеней и возможное отторжение иммунной системой хозяина (Charlesworth et al., 2019).
Стратегии доставки генов можно разделить на три класса: физическая доставка, вирусные векторы и невирусные агенты (рис. 2). Физическая доставка генов может быть осуществлена путем микроинъекции или электропорации. При микроинъекции используется микроскоп и игла диаметром 0,5-5,0 µм Клеточная мембрана прокалывается, и грузы доставляются непосредственно в целевой участок внутри клетки. Этот процесс обходит барьеры, связанные с доставкой через внеклеточные матрицы, клеточные мембраны и цитоплазматические компоненты. В отдельные клетки можно непосредственно вводить либо плазмидную ДНК, кодирующую белок Cas9 и сгРНК, либо мРНК, кодирующую Cas9 и sgRNA, либо белок Cas9 с sgRNA (Horii et al., 2014; Nakagawa et al., 2015). Однако эта стратегия требует значительной подготовки, занимает много времени, и за один раз можно манипулировать только очень ограниченным числом клеток.
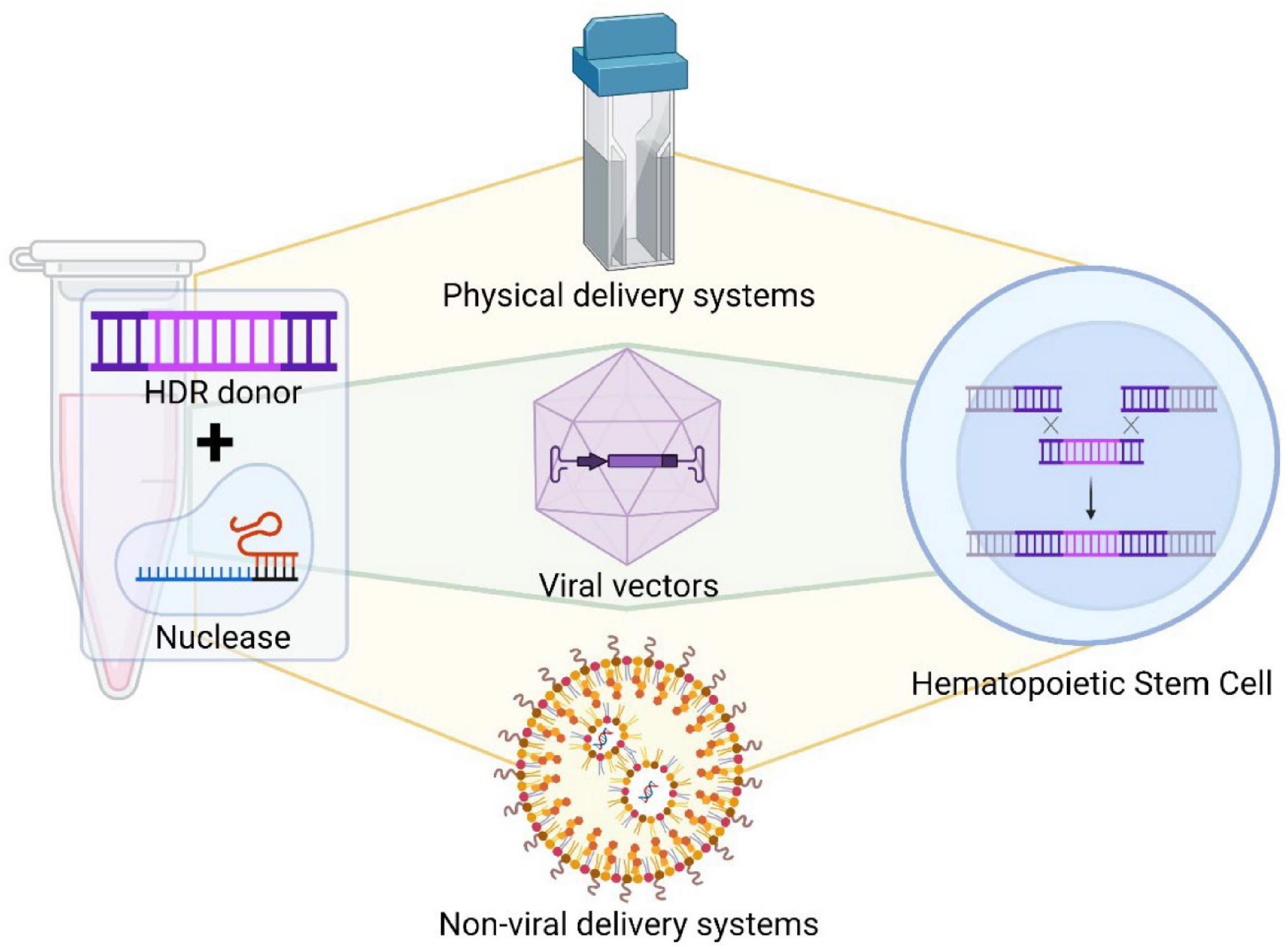
FIGURE 2
Delivery systems of gene editing tools. Different gene editing tools can be delivered into hematopoietic cells by specific delivery systems, such as physical methods, viral vectors or non-viral delivery system. Each gene editing tool has to be modified to be adapted for a specific delivery method. Электропорация (рис. 2) использует импульсные электрические токи высокого напряжения для преходящего открытия пор нанометрового размера в клеточной мембране клеток, что позволяет компонентам проникать в клетку. Электропорация меньше зависит от типа клеток, чем другие методы доставки, и может эффективно переносить груз в клетки, которыми традиционно трудно манипулировать. Существует множество опубликованных методов электропорации клеток млекопитающих (Dumeau et al., 2019), и в настоящее время она широко используется. Однако электропорация может привести к гибели клеток. Для предотвращения этой проблемы были разработаны новые платформы электропорации, позволяющие повысить эффективность доставки и снизить токсичность. Электропорация HSPC может быть эффективной и сохранять свойства приживления и мультипотентности, что облегчает перенос редактирования генов в клинику (Peterson et al., 2016).
Второй метод доставки основан на использовании вирусов (рис. 2). Вирусы эффективно инфицируют клетки и переносят свой генетический материал в организм хозяина, не вызывая иммунного ответа. Этот процесс делает вирусы привлекательными векторами генотерапии. Вирусные векторы были разработаны для переноса желаемого генетического материала и для того, чтобы сделать их более безопасными. Вирусные векторы также неспособны к репликации.
В настоящее время сочетание электропорации для введения нуклеазы и вирусных векторов для доставки донорского шаблона является предпочтительным вариантом для некоторых подходов к редактированию генов (Genovese et al., 2014; Dever et al., 2016; De Ravin et al., 2017; Fananas-Baquero et al., 2021).
Адено-ассоциированные вирусные векторы (AAV) - лучший вариант вектора для доставки донорских шаблонов. AAV представляют собой одноцепочечные ДНК-вирусы, которые широко используются для генотерапии. Известно, что AAV не вызывают и не связаны с какими-либо заболеваниями у людей. Существует также широкий спектр известных серотипов, которые позволяют инфицировать множество клеток с различной специфичностью. AAV серотипа 6 способен эффективно трансдуцировать HSPC в сочетании с электропорацией (Dever et al., 2016; Charlesworth et al., 2018). До 70% HSPC могут быть трансдуцированы без изменения их способности к приживлению. Электропорация нуклеаз в сочетании с rAAV6 для введения большого донорского шаблона была выбрана в качестве очень эффективного метода доставки для генного редактирования HSPC для генотерапии наследственных анемий, таких как гемоглобинопатии (Dever et al., 2016; Hoban et al., 2016; Lattanzi et al., 2021; Wilkinson et al., 2021) и PKD (Fananas-Baquero et al., 2021), а также других заболеваний кроветворной системы, таких как Х-сцепленная хроническая гранулематозная болезнь (X-CGD (De Ravin et al., 2017), синдром Вискотта-Олдрича (WAS) (Rai et al., 2020).
Были изучены и другие платформы вирусных векторов, такие как аденовирусные векторы (HDAdV). Недавно HDAdV продемонстрировали свою способность доставлять редакторы оснований in vivo для ре-экспрессии β-глобина в мышиной модели, несущей дрожжевую искусственную хромосому с локусом человеческого β-глобина (Li et al., 2021).
Кроме того, для HSPC были разработаны другие невирусные системы доставки (рис. 2). Хотя эффективность воздействия на HSPC все еще низка, в ближайшем будущем эти системы cмогут обогнать платформы доставки на основе вирусных векторов, поскольку их легче производить и они более воспроизводимы. Потенциально эти системы также будут более низкими по цене. В настоящее время изучаются различные альтернативы. Некоторые из них, основанные на наночастицах, были использованы для доставки RNP с целью реактивации фетальных глобинов (Cruz et al., 2021). Недавно компания Intellia Therapeutics сообщила об использовании тропных к костному мозгу липидных наночастиц (LNP), способных модифицировать гены в HSPCs in vivo в гуманизированной мышиной модели, в качестве доказательства концепции для лечения SCD (Burns, 2021). Эти невирусные системы начинают становиться эффективными для переноса инструментов редактирования генов в цитоплазму HSPC, поэтому они могут быть легко использованы для редактирования генов с участием только нуклеаз, редакторов оснований или прайм-редакторов. Наконец, были использованы различные подходы для повышения стабильности инструментов редактирования генов внутри клеток, такие как химические модификации (Hendel et al., 2015), добавление альтернативных UTR (Quintana-Bustamante et al., 2019) или содействие ядерной локализации нуклеаз путем связывания с сигналом ядерной локализации (NLS).
Gene Editing Strategies
В настоящее время редактирование генов опирается на нуклеазы. Нуклеазы впервые были использованы для создания DSBs с целью привлечения эндогенного механизма репарации ДНК. В результате появились различные подходы к редактированию генов, основанные на конкретном механизме репарации ДНК: NHEJ, MMEJ и HDR (рис. 3). Кроме того, в настоящее время разрабатываются альтернативные стратегии, основанные на стратегиях, не связанных с DSB, такие как редактирование оснований и редактирование праймеров.
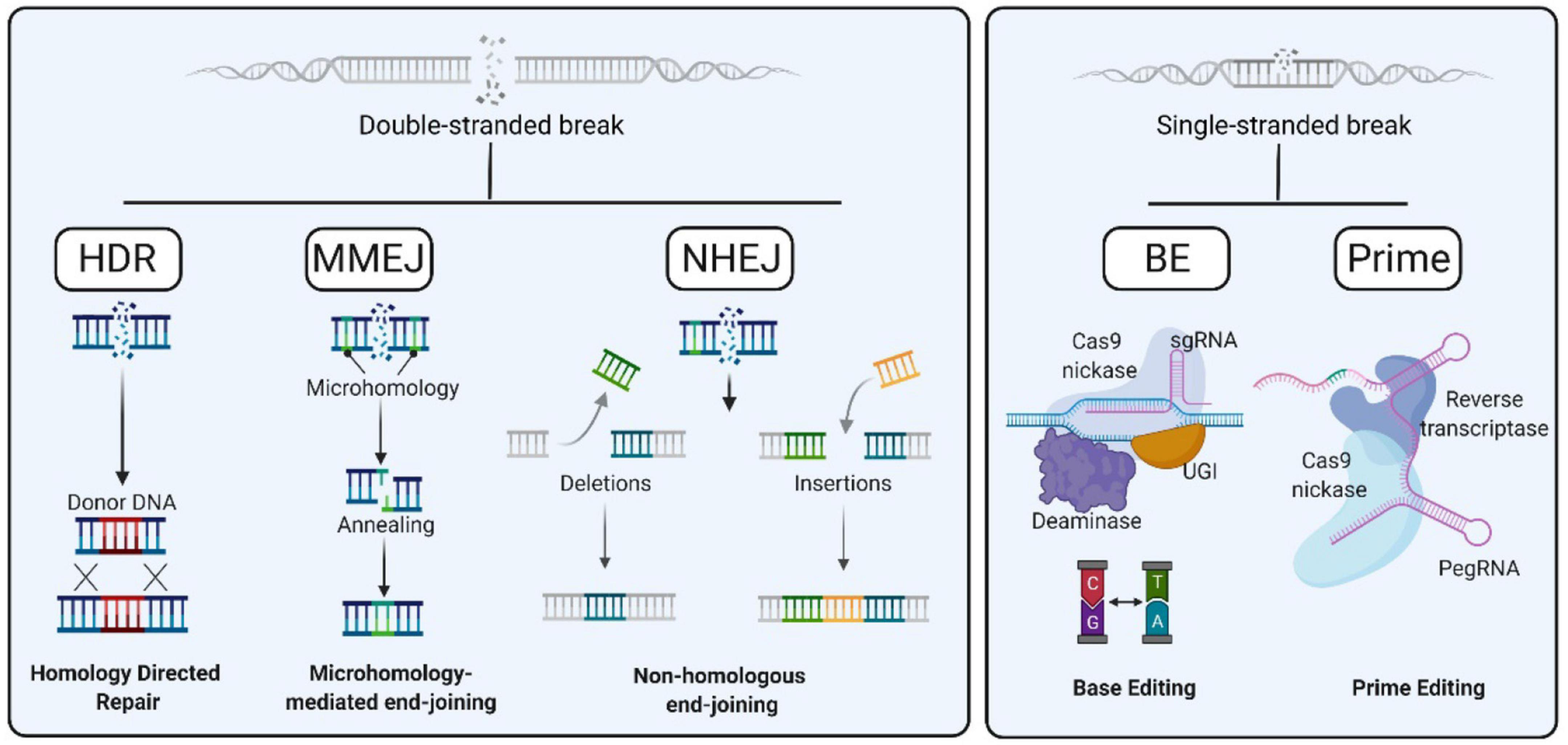
FIGURE 3
Gene editing strategies. After a Double Strand Break (DSBs) event, the preferential cellular pathways for its repair are homology directed repair (HDR), microhomology mediated end joining (MMEJ), and non-homologous end-joining (NHEJ) (left). DNA can also be repaired in a DSB-independent manner, by means of Base Editing and Prime Editing (right). Nonhomologous End Joining-Based Gene Editing
Путь репарации NHEJ является преобладающим способом восстановления DSBs и включает накопление случайных вставок или делеций (indels) в месте разреза (Chang et al., 2017). Он активен во всех фазах клеточного цикла, хотя преимущественно восстанавливает DSBs в фазах G0 и G1 (Shrivastav et al., 2007). Этот механизм подвержен ошибкам и обычно приводит к мутациям со сдвигом рамки, часто создавая преждевременные стоп-кодоны и/или нефункциональный полипептид. Этот путь был особенно полезен в экспериментах по генетическому нокауту и при функциональных геномных экранах CRISPR, но он также может быть полезен в клинике, когда разрушение гена предоставляет терапевтические возможности (Leonova and Gainetdinov, 2020). NHEJ активно исследуется для коррекции гемоглобинопатий. Как упоминалось выше, генерация indels в гене BCL11A, который подавляет фетальные глобины, или в различных позициях в локусе β-глобина, переключает экспрессию глобина взрослых на экспрессию фетального, чтобы компенсировать дефекты этих заболеваний (Canver et al., 2015; Brendel et al., 2016; Ye et al., 2016; Holmes et al., 2017; Lux et al., 2019; Smith et al., 2019; Frangoul et al., 2021).
Обоснование такого подхода заключается в том, что существуют редкие бессимптомные пациенты с SCD, некоторые из которых являются компаунд-гетерозиготными по мутациям SCD и HPFH. У таких пациентов с SCD высокий уровень фетального гемоглобина (HbF) компенсирует их гемоглобинопатию. Молекула гемоглобина представляет собой тетрамер, образованный двумя субъединицами β-подобного глобина и двумя субъединицами α-подобного глобина. Существует последовательное переключение различных пептидов β-подобного глобина, от ε
-глобина в эмбриональном гемоглобине, к γ-глобину в фетальном гемоглобине HbF, а затем к β-глобину в дефинитивном гемоглобине. Этот переход регулируется несколькими активаторами и репрессорами транскрипции, такими как BCL11A, MYB и KLF1. С помощью редактирования генов переключение глобина может быть обращено вспять путем воспроизведения мутаций, таких как HPFH, для улучшения гемоглобинопатии или путем устранения репрессоров фетального глобина, таких как BCL11A. Canver et al. (2015) предприняли первую попытку модулировать переключение глобина путем индуцирования indels в регуляторных областях гена BCL11A. Этим авторам удалось осуществить реактивацию γ-глобина пропорционально уровню редактирования гена эритроидного энхансерного региона BCL11A (Canver et al., 2015). Frangoul et al. (2021) продемонстрировали клиническую пользу нацеливания на BCL11A с помощью системы CRISPR/Cas9 как при SCD, так и при β-талассемии. В настоящее время два пациента проходят лечение с использованием этого подхода, и через год после лечения у них наблюдается повышение фетального гемоглобина, независимость от переливания крови и, в случае SCD, устранение вазоокклюзионных эпизодов (Frangoul et al., 2021). Кроме того, другие группы воспроизвели мутацию HPFH с помощью TALEN или системы CRISPR/Cas9 в качестве альтернативного способа реактивации фетальных глобинов (Ye et al., 2016; Lux et al., 2019).
Homology Directed Repair-Mediated Knock-In
Другой путь, который особенно привлекателен для использования в клинических целях, - это путь безошибочного HR (рис. 3). HR играет важную роль в мейозе и митозе для обмена генетической информацией между сестринскими хроматидами. Он происходит с относительно низкой частотой в клетках млекопитающих, поскольку активен только в фазах S-G2 клеточного цикла (Shrivastav et al., 2007; San Filippo et al., 2008). Этот путь репарации нуждается в гомологичном шаблоне (матрице) ДНК, который может быть использован для программирования модификаций генов. В то время как NHEJ может использоваться для создания нокаутов генов, HR полезен для внесения специфических изменений в сайт-мишень. Этот процесс называется гомологически направленной репарацией (HDR). Для благоприятного протекания HDR экзогенная ДНК, которую предполагается ввести в геном, должна быть фланкирована гомологичными последовательностями, комплементарными области, близкой к DSB. HDR использовалась для введения больших донорских шаблонов с терапевтической кДНК для компенсации функции мутировавшего гена в качестве потенциального лечения гемоглобинопатий (Dever et al., 2016), а также других наследственных заболеваний RBCs, таких как PKD (Fananas-Baquero et al., 2021).
Введение крупного терапевтического донора, как упоминалось ранее, предложило бы глобальное решение для всех пациентов, независимо от природы мутации, которую они несут. Клинически пригодные результаты (до 70% отредактированных HSPC) были получены при использовании системы CRISPR/Cas9 в сочетании с вектором rAAV6 в HSPC для коррекции наследственных гемопоэтических заболеваний, таких как гемоглобинопатии (Dever et al., 2016; Hoban et al., 2016; Lattanzi et al., 2021; Wilkinson et al., 2021), и других заболеваний кроветворения, таких как ase X-CGD (De Ravin et al., 2017), WAS (Rai et al., 2020) и PKD (Fananas-Baquero et al., 2021). В своей фундаментальной работе Dever et al. (2016) описали специфическую коррекцию 50% серповидной мутации в HSPC, полученных от пациентов с SCD. Они сообщили о фенотипической коррекции после дифференцировки этих HSPC в эритроциты путем экспрессии anti-sickling HBB из кДНК терапевтического донора. Эти доклинические исследования свидетельствуют о возможности использования методологии на основе CRISPR для нацеливания в HSPC на глобиновый локус путем HDR, что может быть воплощено в следующем поколении терапии для β-гемоглобинопатий. Недавно мы использовали платформу RNP/rAAV6 для лечения другого заболевания красной крови, такого как PKD (Fananas-Baquero et al., 2021). Мы описали коррекцию PKD путем точного редактирования генов в эндогенном локусе PKLR для поддержания жесткой регуляции фермента RPK во время эритропоэза. Мы показали эффективную коррекцию фенотипа PKD в полученных от пациента клетках HSPC, которые позже дифференцируются в эритроциты. Эритроидные клетки, полученные из отредактированных по гену PKD HSPC, обнаруживают нормальный уровень АТФ (Fananas-Baquero et al., 2021). Это доказательство того, что метаболическая проблема, характерная для PKD, была исправлена путем нокаута кДНК RPK в гене PKLR. Этот подход обеспечит универсальный терапевтический вариант для всех пациентов с PKD, а восстановление RPK будет ограничено эритроидными клетками без изменения остальных кроветворных клеток.
Стоит отметить, что оценки уровня коррекции, полученные при различных заболеваниях эритроцитов SCD, β-талассемии и PKD, выполненные в разных лабораториях, очень похожи, что говорит о воспроизводимости и надежности данной стратегии. Это также позволяет предположить, что ее зрелость может быть распространена на клинические условия.
Single-Stranded Oligonucleotides Based Gene Editing
Введение терапевтического донора является одним из ограничивающих шагов для клинического применения редактирования генов для коррекции наследственных заболеваний кроветворной системы. Чтобы преодолеть это ограничение, некоторые группы применяют высокоспецифичный и персонализированный подход, направленный на исправление точечных мутаций конкретного пациента с минимальным изменением последовательности его генов с помощью таких нуклеаз, как CRISPR/Cas9 и одноцепочечных олигодезоксинуклеотидов (ssODNs). Эти одноцепочечные шаблоны показали свое превосходство над аналогичными шаблонами двухцепочечной ДНК (dsDNA) в плане эффективности HDR, что можно объяснить использованием single-stranded template repair (SSTR), недавно описанного молекулярного механизма репарации ДНК (Gallagher et al., 2020). Кроме того, использование ssODN имеет и другие преимущества, такие как простота конструкции, короткое время производства и более низкая стоимость по сравнению с использованием вирусных векторов. Однако из-за ограничений в длине синтезируемой ssDNA в настоящее время ssODN не используются для вставки длинных последовательностей (Miura et al., 2015). Было предпринято много попыток определить оптимальные характеристики, такие как симметрия, длина гомологичных рукавов или ориентация, которые должны подготовить ssODN для целей редактирования генов, но результаты все еще неясны и зависят от локуса (Richardson et al., 2016).
Возможное применение этой высокоспецифичной системы редактирования генов очень широко, и некоторые заболевания RBCs могут извлечь из нее пользу путем редактирования генов HSPCs ex-vivo, как, например, в случае SCD. Различные группы пытались восстановить ген HBB путем генной коррекции с использованием различных нуклеазных платформ. Hoban и др. ввели ZFNs, нацеленные на мутацию, вызывающую SCD, в виде мРНК путем электропорации вместе с ssODN, несущей корректирующую последовательность (Hoban et al., 2015). Хотя авторы смогли обнаружить тетрамеры гемоглобина дикого типа в эритроидных клетках, полученных из SCD HSPCs, частота коррекции была очень низкой при сравнительном тестировании HSPCs от здоровых доноров. С помощью высокопроизводительного секвенирования ДНК в HSPC здоровых доноров было выявлено до 20% коррекции генов. После трансплантации мышам с иммунодефицитом уровень коррекции генов в костном мозге составил менее 1%. Низкая эффективность редактирования генов в долгосрочных HSPC затруднила его терапевтическое применение (Hoban et al., 2015). Аналогично, DeWitt et al. (2016), Magis et al. (2019) сосредоточились на модификации региона E6V в клетках SCD.
Отредактированные клетки пациентов смогли прижиться у иммунодефицитных мышей, при этом наблюдалось около 2,3% генной коррекции. Это 8-10-кратное общее снижение эффективности от результатов in vitro в HSPCs до ре-популяции in vivo в HSCs согласуется с многочисленными предыдущими отчетами различных групп, занимающихся редактированием генов в HSPCs.
Недавно Pattabhi et al. (2019) напрямую сравнили результаты редактирования генов RNP+rAAV6 с RNP+ssODN при SCD. Исследования in vitro показали, что rAAV6, несущий большой донорский шаблон, способствовал более высокой скорости HDR в отличие от ssODN. Интересно, что уровни коррекции генов in vivo, проанализированные методом секвенирования следующего поколения (NGS), были сходными в обеих группах. Однако при анализе, проведенном через 12 недель после трансплантации, коррекция генов была ниже у мышей, получавших клетки, отредактированные RNP+rAAV6 (около 0,7%), по сравнению с мышами, получавшими клетки, отредактированные RNP+ssODN (до 4%). Этот результат является спорным, поскольку Dever et al. (2016) доказали гораздо более высокую скорость редактирования генов при использовании RNP+rAAV6 для коррекции мутации E6V, даже в долгоживущих HSCs (Dever et al., 2016; Lattanzi et al., 2021). Редактирование генов β-талассемии также было решено с помощью ssODN, с результатами, аналогичными полученным при SCD (Antony et al., 2018).
Помимо гемоглобинопатий, мы попытались использовать стратегию ssODN для коррекции PKD. Мы провели коррекцию четырех мутаций в лимфобластных клеточных линиях, полученных от двух пациентов с PKD, добившись до 20% коррекции генов. Мы наблюдали реверсию функциональности эритроцитов, измеряемую по выработке АТФ, при коррекции HSPCs от пациента с PKD с помощью ssODNs и дифференциации их in vitro в эритроидную линию. Это позволяет рассматривать стратегию CRISPR/Cas9+ssODNs как дополнительную терапевтически значимую систему для пациентов с PKD.
Use of Non-DSB Strategies: Base Editing/Prime Editing
Последние новинки в области редактирования генов - редакторы оснований (BE) и редакторы праймеров (PE) (рис. 3). Обе технологические разработки следуют очень умной стратегии привлечения белков, способных либо преобразовывать один нуклеотид в другой, либо синтезировать новые последовательности ДНК in situ, не требуя расщепления dsDNA или донорской матрицы для HDR. Эти стратегии были впервые разработаны в группе доктора David Liu's , который использовал платформу CRISPR/Cas9 для размещения ферментов, модифицирующих ДНК, в целевом локусе. Сначала были сконструированы редакторы оснований, представляющие собой синтез между нарушенной Cas9 (dCas9 или Cas9 nickase) и ферментом цитидиндеаминазой для прямого преобразования цитозина в уридин, таким образом превращая C в T (Komor et al., 2016; Рисунок 3). Дальнейшее развитие получила система редактирования оснований, позволяющая превращать A-T в G-C путем слияния нарушенного Cas9 с аденозиндезаминазой (Gaudelli et al., 2017). Эта система была названа адениновым редактором оснований (ABE) в отличие от первых цитозиновых редакторов оснований (CBE). Вместе CBE и ABE смогли исправить большинство точечных мутаций человека, вызывающих наследственные заболевания, поскольку на долю переходов C-G -> T-A приходится половина патогенных точечных мутаций. Были внесены различные усовершенствования, которые повысили эффективность систем редакторов оснований и точность их работы. Несмотря на высокую эффективность новейших редакторов оснований, основными проблемами, как и в случае с другими системами редактирования генов, являются внецелевые эффекты, способы предотвращения широкой нуклеотидной интерконверсии и расширение применимости редакторов оснований к любой геномной последовательности. В настоящее время прилагается много усилий для усовершенствования базовых редакторов (Hu et al., 2018). С другой стороны, прайм-редактирование позволяет записывать новую генетическую информацию в определенном целевом геномном локусе путем слияния нарушенного Cas9 с обратной транскриптазой. Prime editing guide RNA (pegRNA) ведет систему прайм-редактирования к нужному локусу, неся при этом генетическую информацию для вставки (Anzalone et al., 2019). PE позволяет осуществлять однонуклеотидные интерконверсии и вставлять несколько нуклеотидов в геномную мишень с очень высокой эффективностью.
Редакторы оснований и прайм-редакторы очень быстро эволюционируют от доказанной концепции до их доклинического применения для коррекции наследственных заболеваний. Было оценено, что редакторы оснований эффективно корректируют SCD (Newby and Liu, 2021). ABE смог преобразовать аллель SCD в непатогенный вариант Makassar. Восемьдесят процентов HSPC, полученных от пациентов, обменивают полиморфизм SCD (CAC) на CGC, присутствующий в Макассарском β-глобине. Когда отредактированные по геному SCD HSPC были пересажены иммунодефицитным мышам, вызванное гипоксией образование серповидноклеточности уменьшилось в три раза. Эта доклиническая работа открывает путь к внедрению редакторов оснований в клинику. Li и др. также описали, как векторы ABE, нацеленные на эритроидный энхансер BCL11A или воссоздающие встречающиеся в природе мутации HPFH в промоторе HBG1/2, могут реактивировать γ-глобин в мышиной модели с помощью хелпер-зависимых аденовирусных векторов, экспрессирующих BE. Более того, in vivo использование редактирования оснований или праймеров может быть легко достижимо, как это было показано для коррекции синдрома прогерии Хатчинсона-Гилфорда (Koblan et al., 2021) или β-гемоглобинопатии (Newby and Liu, 2021). Будущее клиническое применение может быть ускорено тем, что эти новые инструменты редактирования генов не требуют разрезания dsDNA или донорских шаблонных платформ HDR.
Clinical Translation of Gene Editing
Благодаря новым биологическим открытиям область редактирования генов, несомненно, процветает. Высокая эффективность и повышенная безопасность способствовали ее экспоненциальному развитию, и сегодня проводятся первые клинические испытания на основе редактирования генов. На сегодняшний день одобрено более 30 клинических испытаний по редактированию генов, в некоторых из которых использована технология CRISPR/Cas9, и по крайней мере тринадцать из них направлены на коррекцию гемоглобинопатий (см. таблицу 1), что, несомненно, является важной вехой в биомедицинской области.
TABLE 1
Gene editing clinical trials for hemoglobinopathies updated as of December 2021.
С 1980 года многие группы пытались исправить эти патологии с помощью генотерапии с использованием лентивирусных векторов. Это было чрезвычайно сложно, особенно при гемоглобинопатиях, из-за строгих требований к жесткой регуляции глобинов, экспрессия которых ограничена эритроидной линией. Поэтому в лентивирусных конструкциях, разработанных для лечения β-талассемии и SCD, потребовались промоторы, специфичные для данной линии, и в настоящее время они тестируются в нескольких клинических испытаниях (Bueren et al., 2020; Ferrari et al., 2021). Учитывая это ограничение, редактирование генов постулируется как адекватная эволюция генотерапии, способная удовлетворить потребность в физиологическом контроле трансгена.
Как уже упоминалось в этом обзоре, многие группы делают все возможное, чтобы найти терапевтический подход с помощью различных стратегий редактирования генов, таких как HDR или NHEJ. В настоящее время открыто несколько клинических испытаний для оценки возможности аутологичной трансплантации генетически отредактированных HSPC для лечения SCD и β-талассемии после миелоаблативного кондиционирования пациентов (Рисунок 1 и Таблица 1). Девять из этих клинических испытаний основаны на ранее описанной стратегии с обоснованием прерывания регуляторной последовательности эритроидного энхансера гена BCL11A с помощью ZFN, CRISPR/Cas9 или Cas12a. Как упоминалось ранее, эта последовательность участвует в предотвращении активации фетального глобина у взрослых. Эта стратегия может быть применима к пациентам с SCD или с трансфузионно-зависимой (TDT) β-талассемией. В настоящее время открыто шесть многоцентровых клинических испытаний фазы 1/2 для оценки аутологичных продуктов HSPC с генной коррекцией. Два из этих испытаний (NCT03745287 и NCT03655678) оценивают один и тот же препарат на основе CRISPR/Cas9 (CTX001) для лечения SCD и TDT β-талассемии, спонсорами которых являются компании Vertex Pharmaceuticals Inc. и CRISPR Therapeutics. NCT03745287 - это клиническое исследование фазы 1/2/3 с предполагаемым набором 45 пациентов с хорошо документированным тяжелым диагнозом SCD, как минимум двумя тяжелыми вазоокклюзивными кризами в год и правом на аутологичную HSCT. Аналогичным образом, NCT03655678 также является фазой 1/2/3, в которую будут включены 45 пациентов с ТДТ β-талассемии, которые получали не менее 100 мл/кг/год или 10 или более единиц/год переливаний упакованных RBC в течение 2 лет до включения в исследование. Следуя аналогичному подходу, проводится еще одно клиническое исследование, спонсируемое компанией Novartis Pharmaceuticals в сотрудничестве с Intellia Therapeutics для лечения SCD (NCT04443907). Это исследование направлено на оценку безопасности и эффективности двух лекарственных препаратов (OTQ923 и HIX763), которые представляют собой аутологичные HSPC, отредактированные генами CRISPR/Cas9, полученные от пациентов с SCD. Это исследование является клиническим испытанием фазы 1/2, в котором примут участие 30 человек с подтвержденным диагнозом SCD и перенесших как минимум один тяжелый побочный эффект SCD, такой как вазоокклюзионный болевой криз, острый грудной синдром, рецидивирующий приапизм или предшествующий инсульт, а также получавших хронические трансфузии или аллоиммунизацию RBC. Кроме того, в двух других клинических испытаниях (NCT03432364 и NCT03653247) изучается аналогичный подход к редактированию генов, однако этот подход основан на технологии ZFN. Sangamo Therapeutics спонсирует NCT03432364, клиническое испытание фазы 1/2, в котором безопасность, переносимость и эффективность ST-400 (инфузия ZFN-модифицированных аутологичных HSPC) будет оцениваться у 6 пациентов с ТДТ β-талассемией, которым в течение 2 лет до включения в исследование проводилось 8 и более переливаний RBC в год. С другой стороны, компания Bioverativ оценивает безопасность, переносимость и эффективность BIVV003 для аутологичной HSPCT у восьми пациентов с тяжелым SCD.
Недавно были опубликованы первые результаты некоторых из этих клинических испытаний. Первые предварительные результаты NCT03432364, где ST-400 оценивается для ТДТ β-талассемии, не показали положительного результата из-за относительно низкой эффективности редактирования генома, связанной со слабой экспрессией HbF (Smith et al., 2019; Brusson and Miccio, 2021). Однако инфузия CRISPR-отредактированных CD34+ клеток с использованием CTX001 у двух пациентов (один с SCD и один с TDT β -талассемией) (NCT03745287 и NCT03655678) наблюдался высокий уровень аллельного редактирования в костном мозге (до 80% отредактированных клеток) и крови (около 60% отредактированных клеток), общий уровень гемоглобина около 14 g/dL, повышение фетального гемоглобина, независимость от переливания крови и устранение вазоокклюзивных эпизодов у пациента с SCD более чем через год после лечения (Frangoul et al., 2021). Эти многообещающие результаты подтверждают хорошие клинические результаты этой стратегии редактирования генов для лечения SCD и β-талассемии.
Недавно компания Graphite Bio начала новое клиническое исследование фазы 1/2 (NCT04819841) для лечения SCD с помощью другой стратегии редактирования генов, основанной на коррекции мутации E6V с помощью CRISPR/Cas9 и установки rAAV6 (Dever et al., 2016; Lattanzi et al., 2021). Данное исследование является первым на человеке для изучения безопасности и эффективности лекарственного препарата GPH101. Предполагается, что препарат будет протестирован на 15 участниках с диагнозом тяжелой формы SCD, которые также страдают рецидивирующими тяжелыми вазоокклюзивными эпизодами и острым грудным синдромом. По сравнению с другими ранее упомянутыми исследованиями SCD, данное испытание направлено на прямое исправление патогенной серповидной мутации в гене HBB, что позволит сохранить физиологическую регуляцию экспрессии гена. Эта коррекция гена также устранит экспрессию патологического HbS, что решит проблему конкуренции с высоко-экспрессированным патогенным белком (Lattanzi et al., 2021).
Помимо гемоглобинопатий, мы и другие ученые работаем над получением необходимых доклинических данных, которые, скорее всего, в ближайшем будущем перейдут в клинические испытания (Fananas-Baquero et al., 2021). Недавно наша разработка на основе CRISPR/Cas9 и rAAV6 для лечения PKD получила статус орфанного препарата от Европейского агентства по лекарственным средствам (EMA/OD/0000072308), что открывает путь к будущей терапии на основе редактирования генов для пациентов с PKD (рис. 1). Редактирование генов для пациентов с PKD станет шагом вперед в терапии этого заболевания, для которого в настоящее время существуют только паллиативные методы лечения. Несмотря на оценку новых препаратов, таких как аллостерический активатор RPK, Mitapivat (Grace et al., 2019), аутологичная HSCT с отредактированных генами HSPC будет представлять собой окончательную терапию PKD, поскольку одно вмешательство сможет исправить болезнь.
Future Perspectives
Доклинические результаты и ранние клинические данные продемонстрировали возможность генетической коррекции гемопоэтических и не-гемопоэтических наследственных заболеваний с помощью стратегий ex vivo и in vivo (Hoban et al., 2015, 2016; Dever et al., 2016; DeWitt et al., 2016; Ye et al., 2016; De Ravin et al., 2017; Koblan et al., 2021; Li et al., 2021). Современные системы редактирования генов начали переходить в клинику для коррекции наследственных заболеваний РБС. По состоянию на декабрь 2021 года, в настоящее время проводятся 13 клинических испытаний для лечения наследственных заболеваний RBC (Таблица 1). Тремя основными узкими местами этих инновационных методов лечения являются: (i) повышение эффективности для долгосрочного воздействия на HSPC, (ii) оценка безопасности и (iii) облегчение доступа пациентов к терапии редактирования генов. Последний пункт еще более важен в случае гемоглобинопатий из-за большого количества пациентов, страдающих от них. Хотя редактирование генов для лечения заболеваний RBCs уже приносит пользу пациентам с гемоглобинопатиями (Frangoul et al., 2021), эта многообещающая терапия должна быть более доступной для лечения тяжелых пациентов во всем мире.
Имеющиеся инструменты редактирования генов и их использование в различных подходах могут достичь хорошей эффективности при достаточной безопасности в лечении наследственных заболеваний RBC Однако генная терапия будет считаться реальной терапевтической опцией для этих пациентов только тогда, когда производство большого количества адекватно скорректированных клеточных продуктов не будет вызывать затруднений. Это более актуально в случае стратегий, основанных на HDR. Разработка новых модифицированных компонентов, участвующих в процессе HDR, может повысить эффективность HDR и облегчить его потенциальное применение для более широкого спектра генетических заболеваний. В настоящее время тестируются различные стратегии. Базовые редакторы направлены на одноточечные мутации, что потребует одной специфической разработки для каждой генной мутации. Прайм-редактирование направлено на использование более длинных gRNAs, которые несут не только сайт геномного распознавания, но и генетический материал для облегчения исправления мутировавшей последовательности. Экспериментальные данные указывают на совместную локализацию различных компонентов, необходимых для редактирования генов (нуклеазы, донорные шаблоны, дополнительные белки-усилители и т.д.) для облегчения процесса и повышения эффективности. Таким образом, для увеличения потенциального применения терапевтического редактирования генов необходимы новые разработки, способствующие этой субклеточной ко-локализации.
С другой стороны, доставка компонентов редактирования генов в клетку и, что более важно, в клеточное ядро, избегая при этом деградации эндогенными клеточными механизмами на этом пути, является ключевым фактором для редактирования генов. В текущих доклинических разработках (Dever et al., 2016; Schiroli et al., 2019; De Ravin et al., 2021; Fananas-Baquero et al., 2021; Wilkinson et al., 2021) и в недавно одобренных клинических испытаниях для лечения SCD (см. таблицу 1) для доставки рибонуклеопротеина эндонуклеазы и шаблона донорской ДНК используются электропорация и AAV, соответственно. Электропорация обладает присущей ей токсичностью, а AAV требуют очень сложных разработок в производстве и влекут за собой значительные затраты. Для облегчения разработки этих терапевтических стратегий потребуются менее токсичные, более простые и, по возможности, менее дорогие процедуры. Альтернативные системы доставки, такие как наночастицы (Burns, 2021) или вирусоподобные частицы (Mangeot et al., 2019; Hamilton et al., 2021), вместе с новыми донорами (Shy et al., 2021) упростят процесс производства, что сделает их более доступными для большего числа пациентов.
Тем временем, HSCT ex vivo генно-редактированных клеток все еще является ограничивающим фактором для коррекции наследственных заболеваний кроветворной системы. Это еще более актуально для заболеваний, при которых требуемый процент приживления исправленных долгосрочных HSPC высок или когда нет селективного преимущества исправленных клеток над неисправленными. В этом случае требуется режим кондиционирования для устранения эндогенных больных клеток, которые могут конкурировать с исправленными клетками, введенными после процесса редактирования ex vivo. Такие режимы кондиционирования являются высоко генотоксичными и связаны с иммуносупрессией, повреждением тканей и даже стерильностью. Вновь необходимо изучить новые подходы для разработки негенотоксичных режимов кондиционирования, которые позволят приживить достаточное количество исправленных здоровых клеток для достижения терапевтического эффекта.
Кроме того, потенциальное использование методов доставки in vivo облегчит решение проблемы генетических заболеваний, затрагивающих саму кроветворную систему или даже другие органы/системы, которыми невозможно манипулировать для коррекции ex vivo, например, печень, почки, легкие или мозг. Следовательно, разработка новых систем на основе рекомбинантных вирусов или наночастиц, нацеленных на точные ткани или очень четко определенные клеточные популяции, такие как HSPCs, облегчит использование редактирования генов без сложных процессов манипулирования клетками. Однако для снижения побочных эффектов при доставке инструментов редактирования генов в органы пациентов необходимо будет строго контролировать внецелевое воздействие на ткани.
В целом, мы находимся в начале быстро меняющейся области, где постоянно появляются новые инструменты и подходы к редактированию генов. Редактирование генов открывает новые терапевтические возможности для лечения пациентов, страдающих наследственными заболеваниями РБС. Однако сделать эту технологию доступной для большого числа пациентов остается сложной задачей.
