Посещений: 
ЗАВИСИМАЯ ОТ ПЕРЕЛИВАНИЙ КРОВИ β-ТАЛАССЕМИЯ
CRISPR-Cas9 редактирование генов
CRISPR Gene Therapy: A Promising One-Time Therapeutic Approach for Transfusion-Dependent β-Thalassemia—CRISPR-Cas9 Gene Editing for β-Thalassemia Udani Gamage, Kesari Warnakulasuriya, Sonali Hansika and
Gayathri N. Silva
 Thalass. Rep. 2023, 13(1), 51-69; https://doi.org/10.3390/thalassrep13010006ЖУРНАЛ
|
β-Thalassemia is an inherited hematological disorder that results from genetic changes in the β-globin gene, leading to the reduced or absent synthesis of β-globin. For several decades, the only curative treatment option for β-Thalassemia has been allogeneic hematopoietic cell transplantation (allo-HCT). Nonetheless, rapid progress in genome modification technologies holds great potential for treating this disease and will soon change the current standard of care for β-Thalassemia. For instance, the emergence of the CRISPR/Cas9 genome editing platform has opened the door for precision gene editing and can serve as an effective molecular treatment for a multitude of genetic diseases. Investigational studies were carried out to treat β-Thalassemia patients utilizing CRISPR-based CTX001 therapy targeting the fetal hemoglobin silencer BCL11A to restore β-globin expression in place of deficient β-globin. The results of recently carried out clinical trials provide hope of CTX001 being a promising one-time therapeutic option to treat ?-hemoglobinopathies. This review provides an insight into the key scientific steps that led to the development and application of novel CRISPR/Cas9-based gene therapies as a promising therapeutic platform for transfusion-dependent β-Thalassemia (TDT). Despite the resulting ethical, moral, and social challenges, CRISPR provides an excellent treatment option against hemoglobin-associated genetic diseases.

|
Талассемия - это наследственное гематологическое заболевание, которое имеет типичный аутосомно-рецессивный характер наследования [1]. Приблизительно 4,4 из каждых 10 000 живорожденных страдают α- или β-талассемией, что составляет 1,7% населения мира [2,3]. Этот синдром считается опасным для жизни заболеванием в развивающихся странах, где отсутствие ранней диагностики и генетического консультирования способствовало широкому распространению заболевания среди населения [4,5]. α-Талассемия распространена в популяциях Африки и Юго-Восточной Азии, в то время как β-талассемия распространена в странах Средиземноморья, Юго-Восточной Азии, Ближнего Востока, Африки и Южной Америки [6,7]. Эпидемиология талассемии постоянно меняется в связи с миграционными процессами и межрасовыми браками, что приводит к распространению заболевания в регионах, где ранее его не было. Широкое распространение заболевания стало глобальной проблемой здравоохранения и требует новых инновационных методов лечения, которые помогут замедлить его распространение [4].
β-Талассемия классифицируется как талассемия major, minor и intermedia на основании зиготности мутаций гена β-глобина (HBB). Гомозиготная мутация (β0) характеризуется полным отсутствием цепей b-глобина, вызывая большую талассемию или Cooley's анемию [8]. Анемия Cooley's является наиболее тяжелой формой талассемии. Она характеризуется наследованием двух копий дефектного гена β-глобина, что необходимо для возникновения тяжелых клинических признаков заболевания [9]. Дети, наследующие гомозиготные мутации, здоровы при рождении, но основные симптомы заболевания обычно проявляются только через 6 месяцев жизни. На этом этапе развития изменения в транскрипционной регуляции и ремоделировании хроматина вызывают переключение гемоглобина с фетального на взрослый, что приводит к подавлению генов фетального глобина [8,10]. Таким образом, клинические проявления гомозиготных мутаций в HBB становятся очевидными только после завершения процесса переключения [11]. У пациентов с тяжелой формой заболевания отсутствие β-глобина приводит к накоплению непарных цепей β-глобина. Избыток α-глобина агрегирует, образуя преципитаты в эритроидных клетках-предшественниках, повреждая клеточную мембрану и способствуя апоптозу эритроидных клеток-предшественников (неэффективный эритропоэз) [12]. Таким образом, у людей с β-Thalassemia major наблюдается тяжелая анемия, и им приходится переливать кровь в течение всей жизни.
Гетерозиготная мутация (β+) вызывает минорную талассемию, что приводит к бессимптомной анемии со слабой или умеренной микроцитарной анемией. Пациенты, чья клиническая тяжесть находится между талассемией большой и талассемией малой, классифицируются как промежуточная талассемия [13]. Талассемия интермедиа генотипически гетерогенна [11]. Большинство пациентов с промежуточной талассемией являются гомозиготами или сложными гетерозиготами по β-талассемии [9]. В некоторых случаях поражается только один локус β-глобина, в то время как другие локусы полностью нормальны, что приводит к доминантно наследуемой промежуточной талассемии [14,15]. Клинически пациенты с промежуточной талассемией обнаруживают легкую анемию и не нуждаются в пожизненном переливании крови или хелатной терапии [16,17]. Иногда пациенты с талассемией интермедиа подвергаются переливаниям крови, но реже, чем лица с большой формой β-талассемии [11,16].
По последним данным, ежегодно с β-талассемией рождается ~600 000 детей с её симптомами, и приблизительно 80-90 миллионов человек в мире являются носителями этого заболевания [9]. Люди с β-талассемией испытывают серьезные клинические, экономические, психологические и социальные проблемы на протяжении всей своей жизни, поскольку характер стандартных методов лечения тяжелых и умеренных форм талассемии накладывает тяжелое экономическое и психосоциальное бремя на этих пациентов и их семьи [16,18]. Стандартные варианты лечения, широко используемые для лечения β-талассемии, включают переливание крови и хелатную терапию железом [19,20]. Несмотря на широкое клиническое применение, эти методы имеют существенные ограничения и проблемы. Например, повторные переливания крови могут привести к перегрузке железом с опасными для жизни осложнениями, такими как сердечная недостаточность, эндокринная дисфункция и заболевания печени. Напротив, отсутствие адекватного переливания крови приводит к сокращению продолжительности жизни пациентов с трансфузионно-зависимой талассемией (TDT). Таким образом, существует настоятельная необходимость в разработке и тестировании новых терапевтических подходов, которые обещают стать одноразовой терапией для полного излечения заболевания, улучшения выживаемости и качества жизни пациентов с β-талассемией [19,21].
Последние достижения в понимании патофизиологии β-талассемии способствовали разработке новых терапевтических стратегий для этого заболевания. Были разработаны новые подходы к коррекции дисбаланса α- и β-глобиновых цепей для решения проблемы неэффективного эритропоэза и хронической перегрузки железом [19]. Например, фармакологические соединения, такие как гидроксимочевина, которая действует на несколько сигнальных путей для снижения метилирования ДНК в промоторных областях генов γ-глобина для увеличения экспрессии γ-глобина, рекомендуются для лечения β-талассемии. Повышение уровня γ-глобина способствует выработке фетального гемоглобина (HbF) и облегчает тяжесть заболевания, снижая потребность в частых переливаниях крови [22-24]. Более того, гидроксимочевина повышает общий уровень гемоглобина у пациентов и зарекомендовала себя как подходящая стратегия некурабельного лечения при β-талассемии. Гидроксимочевина эффективна при лечении промежуточной формы талассемии, а ее эффективность при лечении большой талассемии еще предстоит подтвердить с помощью рандомизированных клинических исследований [25-27]. Кроме того, рекомбинантный слитый белок Luspatercept (ACE-536), Sotatercept (ACE-011) и соединения, ингибирующие hepcidin и ferroportin , являются одними из новых фармакологических стратегий, направленных на неэффективный эритропоэз для облегчения клинических проявлений заболевания [28-30].
Аллогенная трансплантация гемопоэтических стволовых клеток (HSCT) была единственным эффективным и потенциально излечивающим методом терапии основной формы β-талассемии в течение нескольких десятилетий. При HSCT аллогенные стволовые клетки используются в качестве векторов для коррекции генетических дефектов при β-талассемии путем введения необходимых для нормального кроветворения генов HBB дикого типа [31]. Однако доступ к этой терапии ограничен из-за отсутствия подходящих по антигену лейкоцитов человека (HLA) доноров. После проведения HSCT у пациентов могут развиться долгосрочные серьезные осложнения, включая иммуно-опосредованные заболевания, эндокринные нарушения и нарушение функций легких и дыхательных путей [32-35]. Спленэктомия - еще один метод лечения, который проводится для снижения потребности в переливании крови у пациентов с основной талассемией. Однако она связана с такими неблагоприятными последствиями, как сердечная недостаточность, замедление роста и полового развития, а также повышенная восприимчивость к инфекциям [36].
За последние несколько десятилетий было приложено много усилий для разработки одноразовых лечебных терапевтических подходов для лечения β-талассемии, когда пациент не нуждается в дальнейшем лечении после завершения одного курса лечения. Подходы генотерапии для лечения β-талассемии привлекли значительное внимание как реалистичный и эффективный терапевтический подход для достижения устойчивой, стабильной и высокоуровневой экспрессии функциональных глобиновых генов у пациентов с TDT без смертности и тяжелых иммунологических осложнений, таких как отторжение трансплантата и клональное доминирование [9,34,37,38].
2. Gene Therapy as a Promising Cure for the Acute form of b-Thalassemia
Современные подходы генотерапии предлагают большие терапевтические перспективы с наиболее значительными клиническими результатами при наследственных или приобретенных заболеваниях, включая рак, вирусные инфекции и рецессивные генетические нарушения, такие как талассемия, серповидно-клеточная анемия, муковисцидоз и гемофилия [39,40]. Целью этой методики является исправление дефектных генов путем введения функционального генетического материала в клетки для получения длительного терапевтического эффекта. Стратегии генотерапии можно разделить на две основные категории в зависимости от способа доставки генов, а именно: доставка генов ex vivo и in vivo. Генотерапия ex vivo - это новый подход, который предполагает забор клеток у пациента, их генетическую модификацию в лаборатории и трансплантацию измененных клеток в ткани-мишени. Этот метод имеет большое преимущество перед методом доставки генов in vivo благодаря возможности полностью охарактеризовать и устранить вредные свойства измененных клеток до трансплантации [41]. Пересаженные генетически модифицированные клетки участвуют в секреции и распространении целевых белков в окружающую среду [42]. В отличие от этого, при стратегии in vivo ДНК доставляется непосредственно в резидентные клетки ткани-мишени, обычно с помощью вирусного вектора [40]. Вирусные векторы с дефицитом репликации (например, аденовирусы, аденовирусные ассоциированные вирусы, ретровирусы и лентивирусы) могут функционировать как средства доставки генов для введения генетического материала в клетки-мишени, такие как зародышевые и соматические клетки [43]. В настоящее время эта технология широко применяется в клинических испытаниях для лечения наследственных и приобретенных заболеваний [42].
Вирусные векторы служат популярным методом доставки генов при β-талассемии. Например, лентивирусы были разработаны как отличные кандидаты в векторы для эффективной передачи HBB в моделях человека и животных. В многочисленных моделях мышей и приматов с большой [44] и промежуточной формой [44] талассемии лентивирусный перенос гена β-глобина приводит к уменьшению анемии и последующего повреждения органов, что делает рекомбинантные лентивирусы одной из наиболее эффективных векторных систем для генотерапии β-талассемии [45,46]. Лентивирусные векторы считаются удачным приложением для клинических испытаний благодаря их способности эффективно трансдуцировать клетки, такие как CD34+, которые подвергаются ограниченной пролиферации, а также неделящиеся клетки с повышенной геномной стабильностью [46,47]. В 2006 году было начато клиническое исследование на человеке (исследование LG001) с использованием лентивирусных векторов для лечения β-гемоглобинопатий [47,48]. Лентивирусный вектор 35 HPV569 был использован для переноса гена β-глобина, включающего контрольную область локуса β-глобина (LCR), которая регулирует экспрессию гена β-глобина в аутологичные гемопоэтические стволовые клетки пациентов с β-талассемией [48]. Эта терапия была клинически успешной и устранила потребность в длительном переливании крови через 1 год и сохранялась в течение почти 8 лет. Общий уровень гемоглобина поддерживался на стабильном уровне около 8 g/dL спустя 2-8 лет после лечения. Кроме того, при переливании не было зарегистрировано не-гематологических или связанных с препаратом нежелательных явлений [49].
Недавнее комплексное исследование подтвердило жизнеспособность генотерапии лентиглобином BB305, которая переносит ген βA-T87Q-глобина, кодирующий T87Q аминокислотную замену HbA, в аутологичные CD34+ HSPC. Затем модифицированные ex vivo клетки вливаются обратно пациентам с тяжелой формой талассемии для обеспечения устойчивой экспрессии модифицированного глобина [48,50]. Betibeglogene autotemcel (ZYNTEGLO™), разработанный компанией Bluebird Bio, классифицируется как передовой терапевтический лекарственный препарат (ATMP) и считается первой утвержденной генотерапией, произведенной с использованием лентиглобина BB305 для лечения TDT [51]. Согласно отчету Комитета по оценке рисков Фармаконадзора (PRAC), представленному Европейским агентством по лекарственным средствам (EMA), в июне 2019 года ZYNTEGLO™ получил условное одобрение Европейского союза (ЕС) для клинического лечения пациентов с β-талассемией в возрасте 12 лет и старше, страдающих TDT или не имеющих генотип β0/β0 [52,53]. В клинических исследованиях в рамках исследования North Star на фазах HGB-204, HGB-205, HGB-207 и HGB-212 приняли участие 63 пациента (исследования HGB 207 и HGB 212 все еще продолжаются), в то время как в настоящее время проводится оценка протоколов последующей безопасности для обеспечения долгосрочной стабильности и целостности терапии (исследование LTF-303) [54,55]. ZYNTEGLO™ считается одноразовым генетическим лечением, которое вводит функциональные, сконструированные копии генов β-глобина (βA-T87Q) путем трансдукции аутологичных гемопоэтических стволовых клеток репликационно-некомпетентным, самоинактивирующимся лентивирусным вектором BB305 [56]. Дозировка ZYNTEGLO™ определяется в зависимости от массы тела человека, а введение осуществляется преимущественно путем внутривенной инъекции. Подготовительные клинические исследования продемонстрировали, что модифицированный β-глобин вырабатывается в значительных количествах для поддержания нормального уровня гемоглобина взрослых пациентов, что, возможно, устраняет необходимость в переливании эритроцитов. Европейская комиссия (EC) определила этот препарат для лечения средней и тяжелой формы β-талассемии в качестве orphan фармацевтического продукта. Управление по контролю за продуктами и лекарствами США (FDA) также предоставило ZYNTEGLO™ статус orphan препарата для лечения TDT [53].
Хотя лентивирусные векторы относятся к платформе вирусных векторов на основе РНК, они могут быть модифицированы для уменьшения их патогенности. В настоящее время проводятся клинические исследования для оценки возможного применения лентивирусных векторов третьего поколения и их инженерии для эффективного переноса генетического материала в клетки-мишени при сохранении долгосрочной стабильной экспрессии [57]. Инженерия само-инактивирующихся лентивирусных векторов проявляется в удалении вирусных длинных терминальных повторов, что приводит к увеличению грузовой емкости, которая может вмещать измененный целевой ген и его регуляторные элементы для усиления экспрессии трансгена [44]. Более того, не-клинические исследования на мышиных моделях выявили незначительные угрозы генотоксического эффекта из-за трансактивации генетических элементов после переноса лентивирусных векторов, несущих ген человеческого β-глобина [58]. Однако, согласно руководящим принципам, принятым такими регулирующими органами, как FDA и EMA, для безопасности и эффективности генотерапии лентиглобином необходимо тщательное изучение определенных параметров, таких как токсичность вектора и стабильная интеграция [59]. В феврале 2021 года появились опасения после оценки пациента, получившего генную терапию лентиглобином, у которого была диагностирована острая миелоидная лейкемия (AML) [60]. Хотя имеющиеся данные не указывают на инсерционный мутагенез и риск образования опухоли при генотерапии ZYNTEGLO™, PRAC рекомендует частое (не реже одного раза в год) наблюдение за пациентами на предмет неблагоприятных состояний, таких как миелодисплазия, лейкемия или лимфома [55].
Хотя вирусные методы доставки генов позволяют достичь более высокой эффективности трансфекции, их применение ограничено из-за проблем безопасности и токсичности. Например, иммуногенность вектора, возможность онкогенной трансформации и внезапная смерть реципиентов привели к замене лечения на основе вирусных векторов альтернативными стратегиями генотерапии [61,62]. Появление инструмента для редактирования генома Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated protein 9 (CRISPR/Cas9) предлагает осуществимый и элегантный вариант редактирования генов, позволяющий безопасным и эффективным образом вносить желаемые генные модификации, снимая некоторые трудности, связанные с традиционной генной терапией [63]. Недавно предварительные результаты клинического испытания CRISPR-фазы-1 возродили надежду на возможное прорывное одноразовое лечение β-талассемии [64].
3. Advances in CRISPR Gene Therapy Hold Great Promise as an Effective One-Time Treatment Option for TDT
CRISPR - это прорывная технология редактирования генома с помощью РНК в 21 веке, которая открывает большие терапевтические перспективы против наследственных заболеваний крови [21]. CRISPR-опосредованные адаптивные иммунные системы были впервые обнаружены у бактерий, которые обеспечивают защиту от вторжения чужеродных генетических элементов [65-68]. Бактериальный геном содержит короткие повторяющиеся последовательности, известные как массивы CRISPR, разделенные неповторяющимися протоспейсерными последовательностями, которые будут транскрибироваться и перерабатываться в зрелые последовательности CRISPR РНК (crRNA), гомологичные вторгшемуся генетическому материалу. Сборка активного эффекторного комплекса CRISPR/Cas требует включения зрелой crRNA, которая направляет эффекторный комплекс к вторгающимся нуклеиновым кислотам, и отдельно кодируемой многодоменной нуклеазы Cas (нуклеаза класса I) или набора нуклеаз Cas (нуклеазы класса II) [67]. Распознавание мишени происходит благодаря обширной комплементарности между последовательностью ДНК захватчика и crRNA, что приводит к Cas-зависимому двухцепочечному расщеплению комплекса crRNA-чужая ДНК [65,69]. Система CRISPR типа II, состоящая из одной эффекторной нуклеазы Cas9, является самой простой и хорошо изученной системой CRISPR, которая широко применяется в геномной инженерии [65,66,69]. Система типа II требует наличия второй молекулы РНК, называемой транс-активирующей crRNA (tracrRNA), которая соединяется с crRNA для образования двойной структуры guide RNA (gRNA), необходимой для распознавания мишени и crRNA-guided расщепления ДНК, предшествующей protospacer adjacent motif (PAM), присутствующему в последовательности мишени [67,70,71]. Замечательная способность систем типа II к программированию была использована для разработки мощных инструментов редактирования генов, позволяющих целенаправленно воздействовать на любую желаемую последовательность ДНК путем настройки последовательности crRNA. Хотя в природе crRNA и tracrRNA кодируются отдельно, ученые создали химерную молекулу однонаправленной РНК (sgRNA) путем слияния crRNA с tracrRNA. В терапевтических подходах на основе CRISPR важным моментом является безопасная и эффективная доставка системы CRISPR/Cas9 в клетки-мишени. Система CRISPR/Cas9 может быть введена в изолированные клетки-мишени пациентов с β-талассемией с помощью вирусных или невирусных векторов доставки, где РНК-зависимое расщепление ДНК осуществляется Cas9 после распознавания целевой последовательности crRNA. Электропорация и микроинъекция могут быть указаны как безопасные невирусные методы доставки CRISPR [39,72,73].
Глубокое понимание патофизиологии β-талассемии привело к успешному применению инструмента редактирования генома CRISPR/Cas9 от стендовых до клинических испытаний для использования в качестве перспективного лечебного терапевтического вмешательства при β-талассемии. Одним из широко изученных подходов генотерапии этого заболевания является восстановление нормальной экспрессии β-глобина путем исправления вызывающих болезнь мутаций HBB с помощью CRISPR/Cas9-опосредованной гомологически-направленной репарации (HDR) [11,21]. Тем не менее, этот подход затруднен наличием широкого спектра мутаций в HBB, поскольку для использования в терапии необходимо разработать, оптимизировать и утвердить специфические sgRNA и донорные шаблоны для каждой мутации HBB. Однако ученые успешно применили инструмент CRISPR/Cas9 для исправления мутаций HBB в полученных от пациента индуцированных плюрипотентных стволовых клетках (iPSCs) [74]. Исследование, направленное на исправление двух различных мутаций HBB (кодон 41/42 с делецией 4 п.н. (-TCTT) и -28 с заменой (A > G) в промоторе) с помощью CRISPR/Cas9-опосредованного HDR привело к переходу гомозиготной ?-талассемии в гетерозиготное состояние и восстановлению нормальной экспрессии HBB в эритроцитах, дифференцированных из исправленных iPSCs [75]. Генетические исправления в HBB с использованием того же подхода наблюдались в ряде других исследований [75-79]. Например, инструмент CRISPR/Cas9 и донорский шаблон ssODN (одноцепочечный олигодезоксинуклеотид) были использованы для генетической коррекции iPSCs, полученных от пациента, дважды гетерозиготного по гемоглобину E/β-талассемии. Затем исправленные клоны были дифференцированы в эритроидные клетки с восстановленной экспрессией зрелого β-глобина [77]. В аналогичном исследовании инструмент CRISPR/Cas9 был использован для коррекции iPSCs с ?-талассемией с гомозиготной мутацией CD17 (A > T) в локусе HBB. Исправление мутации привело к восстановлению нормальной экспрессии β-глобина с улучшением способности к кроветворной дифференцировке и устранению неэффективного эритропоэза [78,80]. Более того, исследование in vivo продемонстрировало успешную коррекцию мутации HBB (гомозиготная делеция 41/42) в iPSCs, специфичных для пациента, путем восстановления экспрессии β-глобина в дифференцированных HSCs без опухолевого генеза, что подтверждает безопасное клиническое применение генотерапии CRISPR/Cas9 для лечения β-талассемии [81].
В новых стратегиях лечения TDT значительное внимание уделяется молекулярным механизмам переключения глобулинов для постоянного восстановления баланса α- и β-глобиновых цепей у пациентов [82,83]. Технология CRISPR была успешно применена для реактивации ранее подавленного гена γ-глобина с целью увеличения выработки HbF у пациентов с TDT [7,82]. На стадии плода синтез γ-глобина уравновешивает уровень α-глобина, объединяясь для образования адекватного уровня HbF (α2γ2), в то время как синтез β-глобина замещает γ-глобин для образования гемоглобина взрослых (HbA, α2β2) после рождения (в течение первого года жизни), который преобладает во взрослой жизни [83]. Во взрослых ретикулоцитах экспрессия гена γ-глобина заглушается геморегуляторным ингибитором (HRI, также известным как EIF2AK). HRI - это эритроид-специфическая киназа, которая может фосфорилировать фактор инициации трансляции eIF2α Фосфорилирование eIF2α ингибирует трансляцию мРНК γ-глобина с одновременным снижением уровня HbF у взрослых [21,84]. Транскрипционный фактор с цинковымb пальчиками BCL11A (B cell lymphoma/leukemia 11A) считается критическим посредником в подавлении экспрессии γ-глобина и снижении уровня HbF в эритроидных клетках взрослого человека во время переключения с γ- на β-глобин. BCL11A работает как транскрипционный репрессор экспрессии γ-глобина через прямое связывание BCL11A с мотивом TGACCA в промоторе γ-глобина, заглушая выработку HbF во время переключения гемоглобина с фетального на взрослый [85,86,87] (Рисунок 1a).
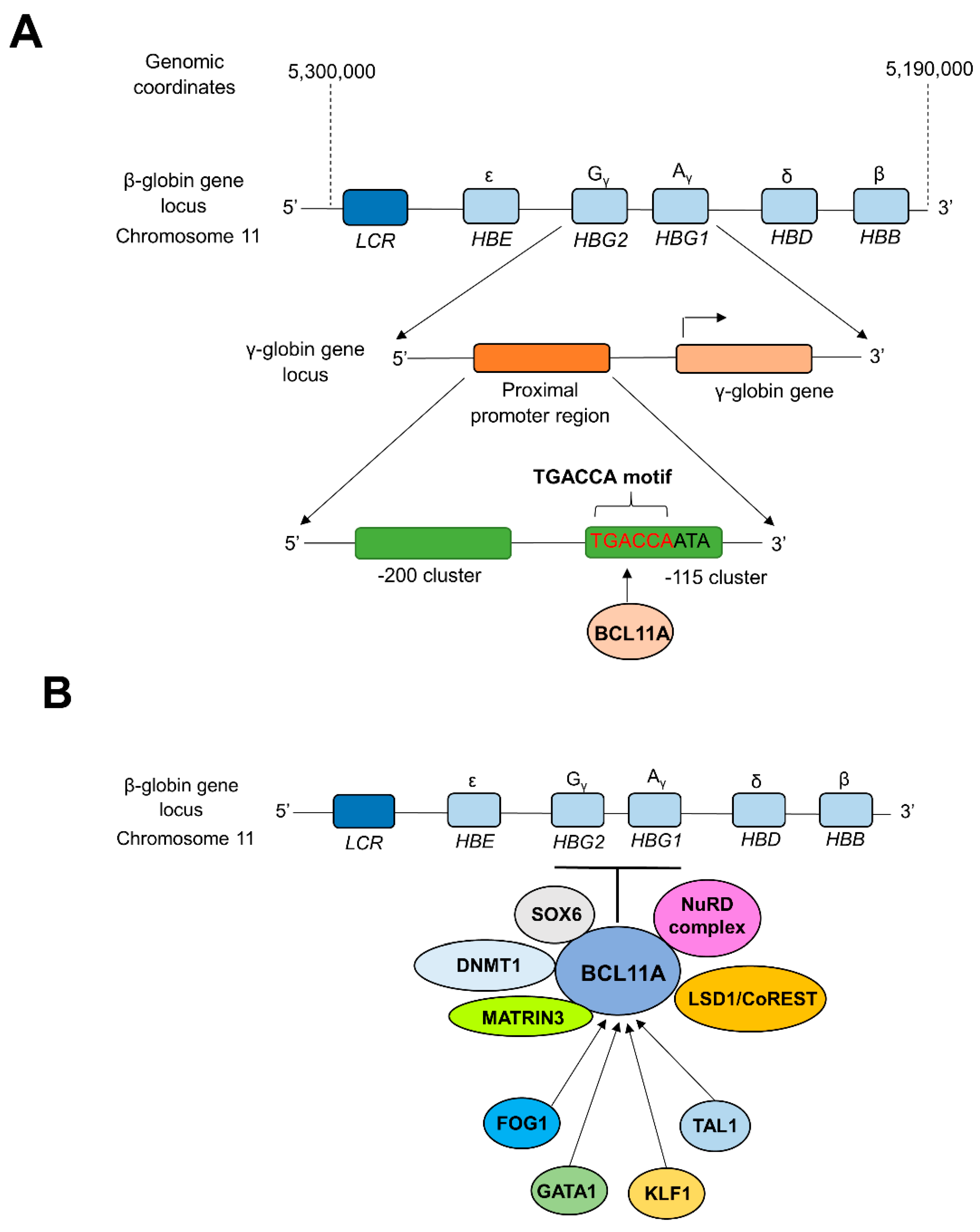 Figure 1. BCL11A-mediated transcriptional silencing of β-globin. (A) Direct repression of the promoter region of β-globinhttps://pub.mdpi-res.com/thalassrep/thalassrep-13-00006/article_deploy/html/images/thalassrep-13-00006-g001.png?1675670339 genes by BCL11A. Interactions between zinc finger motifs of BCL11A and the proximal TGACCA motif (-118 to -113) of β-globin promoter silence the expression of β-globin during fetal to adult hemoglobin switching [86,89]. (B) Long-range inthttps://pub.mdpi-res.com/thalassrep/thalassrep-13-00006/article_deploy/html/images/thalassrep-13-00006-g001.png?1675670339eractions of BCL11A with multi-protein complexes and transcription factors. The level of expression of BCL11A is controlled by several transcription factors such as FOG1, GATA1, TAL1, etc. At the same time, BCL11A is capable of repressing the β-globin expression through long-range interactions and cooperation with multi-protein complexes such as NuRD, DNMT1, MATRIN3, etc. [90,91,92]. LCR; β-globin locus control region, HBE; Hemoglobin Subunit Epsilon gene, HBG1; Hemoglobin subunit Gamma 1 gene, HBG2; Hemoglobin Subunit Gamma 2 gene, HBD; Hemoglobin Subunit Delta gene, HBA; Hemoglobin Subunit Alpha gene. NuRD; Nucleosome Remodeling and Deacetylase, LSD1/CoREST; Lysine Specific Demethylase 1/Repressor Element 1 Silencing Transcription Factor Corepressor 1, MATRIN3; Nuclear Matrix Protein-3, DNMT1; DNA Methyltransferase 1, SOX6; SRY-box transcription factor 6, TAL1; T-cell Acute Lymphoblastic Leukemia 1, KLF1; Kruppel-Like Factor 1, GATA1; GATA binding protein 1, FOG1; Zinc finger protein FOG family member 1.
Установлено, что снижение уровня BCL11A уменьшает выработку HRI, что в конечном итоге усиливает выработку HbF [84]. Более того, встречающиеся в природе мутации в мотиве TGACCA связаны с высокой персистенцией HbF. Основываясь на этих данных, ученые предположили, что изменений в мотиве TGACCA будет достаточно, чтобы реактивировать производство HbF на достаточном уровне для смягчения тяжести β-талассемии [88]. Редактирование гемопоэтических стволовых клеток ex vivo и in vivo проводилось с использованием трансгенных мышей, несущих локус человеческого β-глобина (β-YAC) для ингибирования связывания BCL11A с мотивом TGACCA в промоторе γ-глобина с целью увеличения экспрессии γ-глобина [87]. В данном исследовании был использован хелпер-зависимый аденовирусный вектор человека CD46, включающий компоненты CRISPR/Cas9 для разрушения области связывания BCL11A в промоторе γ-глобина. Это исследование продемонстрировало эффективное расщепление целевого локуса, что привело к поразительному переходу от экспрессии β- к экспрессии γ-глобина без каких-либо гематологических отклонений [88].
Другой механизм, используемый BCL11A для подавления экспрессии γ-глобина, включает связывание с промотором γ-глобина через дальние взаимодействия с мультибелковыми комплексами (Рисунок 1b). BCL11A-содержащие многосубъединичные комплексы обычно содержат различные транскрипционные ко-репрессоры и хроматин-модифицирующие субъединицы (например, лизин-специфическую деметилазу 1 и repressor element-1 silencing transcription factor corepressor 1 complex (LSD1/CoREST demethylase complex), ДНК-метилтрансферазу 1 (DNMT1) и нуклеосомный ремоделирующий, деацетилазный комплекс (NuRD), и SRY-Box transcription factor 6 (SOX6)). Например, SOX6 репрессирует экспрессию гена γ-глобина, связываясь с проксимальными частями промотора γ-глобина, чтобы облегчить привлечение BCL11A к его сайту связывания через дальние меж-белковые взаимодействия [91,93]. Введение CRISPR/Cas9-опосредованных мутаций indel в консенсус SOX6 в промоторе γ-глобина предотвращало взаимодействие SOX6-γ-глобинового промотора, следовательно, реактивировало экспрессию гена γ-глобина. CRISPR-опосредованное включение трех различных indels в трех различных локусах в консенсусе SOX6 в промоторе глобина предотвратило взаимодействие промотора SOX6-γ-глобина в клетках K562. Эти CRISPR-модифицированные клетки с процентом indels 30%, 25% и 24% показали 1,3, 2,1 и 1,1 кратное увеличение экспрессии гена γ-глобина, соответственно [94].
Экспрессия BCL11A транскрипционно регулируется путем связывания транскрипционных активаторов с эритроид-специфическим энхансерным регионом BCL11A. GATA-1 - это транскрипционный активатор с цинковыми пальчиками, который специфически связывается с последовательностью связывания транскрипционного активатора GATA-1 (мотив GATA) в эритроид-специфической области энхансера BCL11A для усиления его экспрессии [95] (Рисунок 2).
 Figure 2. Schematic depiction of the transcription factor binding sites in the erythroid enhancer region of BCL11A. The binding of transcription factors (GATA1, TAL1) increases the BCL11A expression resulting in decreased β-globin gene expression. Disruption of the erythroid-specific enhancer of BCL11A increased the endogenous expression of β-globin and HbF production in adults. This approach is identified as an effective therapeutic strategy to treat β-hemoglobin disorders [96]. GATA1; GATA binding protein 1, TAL1; T-cell acute lymphoblastic leukemia 1. The major transcriptional activators of BCL11A, GATA1, and TAL1 bind to the erythroid enhancer elements in intron 2.
GATA-1 также сотрудничает с BCL11A в качестве компонента комплекса NuRD во время переключения с γ- на β-глобин [97]. Поэтому нарушение эритроид-специфической области энхансера BCL11A для снижения его экспрессии рассматривается как эффективный терапевтический подход к лечению β-гемоглобинопатий. CRISPR/Cas9-опосредованное удаление 200 п.н. в эритроид-специфической области энхансера [96] BCL11A привело к снижению экспрессии BCL11A и реактивации гена γ-глобина в клетках K562 [98]. В другом исследовании электропорация Cas9 и crRNA длиной 20 нт, нацеленных на сайт связывания GATA-1 в эритроидном энхансере +58 BCL11A в CD34+ HSPCs, привела к высоко-проникающему нарушению этого мотива, что привело к снижению экспрессии транскрипта BCL11A на 54,6% и одновременному повышению экспрессии γ-глобина на 35,3-75,1% по сравнению с 14,7% в не-модифицированных клетках [99,100]. Недавно была проведена аналогичная работа по созданию indels в этом мотиве с помощью нуклеаз с цинковыми пальцами (ZFN). Исследовательская клеточная терапия ST-400, состоящая из ex vivo, ZFN-опосредованного разрушения сайта связывания GATA BCL11A в аутологичных CD34+ клетках, в настоящее время проходит фазу 1/2a клинических испытаний для повышения уровня HbF в CD34+ клетках от пациентов с β-талассемией и человеческих эритроидных клетках взрослой стадии. Ожидается, что исследования безопасности и эффективности будут проводиться в течение 3 лет после введения. Сообщалось о серьезных нежелательных явлениях, связанных с гиперчувствительностью, но с ними успешно справились с помощью медицинского вмешательства [101].
Промоторная область BCL11A или его энхансер-связывающий сайт могут служить прекрасными мишенями для ингибирования или понижения экспрессии BCL11A. Несколько исследований продемонстрировали отличные перспективы целенаправленного разрушения эритроид-специфической области энхансера BCL11A с помощью системы CRISPR/Cas9, что приводит к значительному повышению уровня γ-глобина [99,100,102]. Используя продуктивность этой стратегии, спонсируемые промышленностью клинические испытания проложили путь к оказанию передовой медицинской помощи при β- гемоглобинопатиях.
4. CTX001 Therapy
CTX001 - это исследовательская генетически модифицированная клеточная терапия, изучаемая компаниями CRISPR therapeutics (Кембридж, штат Массачусетс, США) и Vertex Pharmaceuticals (Бостон, штат Массачусетс, США) для лечения наследственных гематологических заболеваний, таких как серповидно-клеточная болезнь (SCD) и TDT. В настоящее время клинические испытания CTX001 набирают пациентов из США. Это невирусное, ex vivo CRISPR/Cas9-опосредованное редактирование сайта специфического редактирования в эритроид-специфическом сайте связывания усилителя BCL11A в аутологичных CD34+ гемопоэтических стволовых и клетках предшественниках (HSPC) для увеличения экспрессии гена γ-глобина и, впоследствии, повышения уровня HbF у пациентов. Было показано, что введение CTX001 приводит к изменению 80% аллелей в локусе BCL11A без признаков внецелевого редактирования [64]. Повышение уровня HbF снизило потребность в переливании крови и анемическое состояние при β-талассемии, а также уменьшило клинические осложнения у пациентов с SCD, такие как вазоокклюзивные кризы (VOCs) [103].
Первыми клиническими исследованиями CTX001 на людях для лечения SCD и TDT являются CLIMB SCD-121 (NCT03745287) и CLIMB THAL-111 (NCT03655678), соответственно [103]. CLIMB SCD-121 - это фаза 1/2, dj многих местах, открытое исследование с одной дозой, в котором изучается безопасность и эффективность CRISPR/Cas9-модифицированных аутологичных CD34+ HSPC у пациентов с SCD. К участию в исследовании допускались пациенты в возрасте от 12 до 35 лет с тяжелой формой SCD с историей по крайней мере двух VOCs в год в течение предыдущих двух лет [104]. Безопасность и эффективность исследования контролировались в течение от шести месяцев до двух лет после вливания CTX001. Предварительные результаты терапии показали, что пациенты были свободны от VOCs, и у пациентов были обнаружены клинически значимые уровни HbF при отсутствии серьезных нежелательных явлений, связанных с CTX001 [103,105] (Таблица 1).
Table 1. Details of the pivotal clinical trials utilizing CRISPR/Cas9 gene therapy for β-hemoglobinopathies.
Продвижение терапии CTX001 создало многообещающий терапевтический подход для лечения β-талассемии. Ранее право на участие в программе CLIMB THAL-111 было ограничено пациентами в возрасте 18-35 лет, но позже оно было изменено на пациентов в возрасте 12-35 лет с установленной историей переливаний законсервированных эритроцитов в объеме не менее 100 мл/кг массы тела/год в течение предыдущих двух лет [104]. CD34+ HSPC были собраны у пациентов путем мобилизации с помощью filgrastim и plerixafor с последующим аферезом. Затем эти клетки были модифицированы с помощью редактирования генома, опосредованного CRISPR/Cas9. Здесь использовалась sgRNA длиной 20 нт, нацеленная на область эритроидного энхансера BCL11A, а Cas9 выполнял сайт-специфическую эндонуклеазную активность, что привело к успешному разрушению этого мотива. После тестирования и контроля качества модифицированные клетки были направлены на инфузию. После миелоаблативного кондиционирования бусульфаном пациенты получали CRISPR-модифицированные CD34+ HSPC в виде внутривенного однократного вливания (рис. 3). Этих пациентов контролировали на предмет приживления, восстановления кроветворной системы и нежелательных явлений наряду с повышением уровня HbF и общего Hb [64,103,104] (Таблица 1).
 Figure 3. CTX001 therapy workflow. CTX001therapy is a potential one-time gene therapy for TDT and severe SCD that relies on CRISPR/Cas9 gene editing to facilitate the patient's own cells to produce high levels of HbF. CRISPR/Cas9 system targeting the deletion of erythroid-specific enhancer binding site of BCL11A is introduced into CD34+ stem cells from β-Thalassemia patients, resulting in downregulation of BCL11A expression. Subsequent inactivation of BCL11A binding to the Pβ-globin causes reactivation of the β-globin gene, resulting in fetal globin switch. After quality controlling and clinical testing, CRISPR-modified CD34+ stem cells are introduced into β-Thalassemia patient via autologous transplantation, along with myeloablative chemotherapy and immune-suppressive treatments [103,104,105]. PBCL11A; Promoter region of BCL11A gene. * (Asterisk symbol); Indel mutation introduced by CRISPR/Cas9.
Терапия CTX001 привела к нарушению эритроид-специфического сайта связывания энхансера BCL11A, что привело к подавлению экспрессии BCL11A. В результате транскрипционное глушение γ-глобина BCL11A отменяется, и экспрессия γ-глобина реактивируется, повышая уровни HbF и общего Hb у пациентов с TDT, получавших CTX001. Сохранение повышенных уровней HbF и общего Hb также наблюдалось у пациентов с течением времени [105,111]. В течение 21,5 месяца после получения терапии CTX001 у самого первого пациента было отмечено 32 нежелательных явления, но они находились на ранних стадиях тяжести. У некоторых пациентов в ранних клинических испытаниях наблюдались тяжелые нежелательные явления, такие как гемофагоцитарный лимфогистиоцитоз и острый респираторный дистресс-синдром [64,103]. В целом, было установлено, что эти неблагоприятные явления коррелируют с миелоаблацией бусульфаном и аутологичной трансплантацией HSPC [104]. Другие наблюдаемые негативные эффекты включали пневмонию и сепсис с нейтропенией, вено-окклюзионную болезнь и синдром обструкции синусоидов, абдоминальный дискомфорт и желчекаменную болезнь, а также несерьезные последствия лимфопении (Таблица 1) [64]. Возникновение этих эффектов может быть связано с задержкой восстановления лимфоцитов из-за обогащения CD4+ Т-клетками после терапии CTX001 [64,112]. Исследование CLIMB-Thal-111 было предназначено для оценки безопасности и эффективности дозирования CTX001 для пациентов в возрасте 12-35 лет при одобрении протоколов длительного наблюдения до 15 лет (CLIMB-Thal-131) [113].
В дополнение к испытаниям CTX001, ST-400 - это еще один тип исследуемой аутологичной клеточной терапии ex vivo, в которой используется редактирование генов для увеличения производства HbF. Это клиническое исследование фазы 1/2, в котором используется технология Zinc Finger Nuclease (ZFN) компании Sangamo, способствующая целенаправленному редактированию генома. Испытания ST-400 проводятся по схожей с CTX001 стратегии, направленной на разрушение усилителя гена BCL11A с последующей реактивацией экспрессии гена γ-глобина [114]. Тем не менее, продолжающиеся испытания CTX001 с положительными результатами свидетельствуют о том, что эта новая терапия обладает значительным потенциалом для использования в лечении пациентов с TDT [44]. Однако в ходе клинического применения большое внимание должно уделяться таким побочным явлениям, таким как непреднамеренное внецелевое редактирование, неэффективная доставка генов и аутоиммунные реакции. Следовательно, тщательный мониторинг и наблюдение за клинической эффективностью, генотоксичностью и другими протоколами безопасности являются обязательными. Для определения риска наблюдения возможных неблагоприятных событий необходимо использовать комплексные доклинические исследования, лабораторные и вычислительные методы [64]. Дальнейший прогресс в экспериментах и понимание лежащих в основе молекулярных концепций безопасности и эффективности терапии CTX001 прольет свет на терапию, улучшая качество жизни пациентов.
5. Relative Merits
Технология редактирования генов, опосредованная CRISPR, привела к трансформации и созданию перспективного метода лечения TDT. Этот подход позволяет избежать трудоемких этапов белковой инженерии, таких как изменение и генерирование сайт-специфических нуклеаз, распознающих определенную последовательность ДНК [115], одновременно повышая простоту дизайна мишени и сокращая время реализации. Поэтому CRISPR/Cas9 зарекомендовал себя как простой, эффективный и рентабельный метод генотерапии наследственных заболеваний крови [116]. В отличие от опосредованной вирусными векторами вставки генов, редактирование генома с помощью CRISPR не требует использования интегрирующих векторов, поскольку транзиторное производство специфической эндонуклеазы способно вызвать необходимое расщепление ДНК.
Несмотря на то, что технология CRISPR устраняет некоторые препятствия, связанные с традиционной генотерапией, она имеет некоторые недостатки в клинических условиях. Основной проблемой этого метода является высокая частота непреднамеренных расщеплений генома, что приводит к внецелевому мутагенезу, вызывающему непредвиденные и нежелательные последствия во время терапевтических испытаний. Внецелевые эффекты представляют опасность для жизни из-за пагубных генетических изменений и нарушения функции генов, что в конечном итоге приводит к неблагоприятным фенотипам. В настоящее время исследователи находятся в процессе изучения различных стратегий для смягчения этой проблемы [73,117]. Внецелевые эффекты оценивались с помощью различных методик в ходе CRISPR-генной терапии при β-талассемии [118]. Несколько исследований показали, что использование высокоточных ферментов и стратегии двойного связывания может значительно снизить внецелевую активность при сохранении приемлемой эффективности редактирования генома-мишени [118,119]. Например, недавно для iPSC пациента с мутацией β-талассемии (IVSII-1 G > A) была применена эффективная стратегия целенаправленного воздействия на гены с помощью двойной никазы Cas9 путем интеграции каталитической мутантной никазы Cas9D10A с парой sgРНК, комплементарных целевой последовательности [119,120]. Оптимизация одиночных направляющих РНК также может повысить эффективность и точность редактирования генов [117,121]. Применение методов in silico для предсказания и количественной оценки эффектов вне мишеней важно для преодоления этой проблемы [117,121,122].
Однако привлекательное использование этого подхода ставит определенные проблемы этического, социального характера и безопасности, особенно при использовании его в клинической практике [123]. Неопределенность в отношении того, вызывает ли такая терапия необратимые генетические изменения в организме и передается ли измененный ген последующим поколениям, вызывает серьезную озабоченность. Даже если геном будет изменен в соответствии с планом и намеченная функциональная проблема будет решена в указанное время, неясно, будет ли четко определено сложное взаимодействие между генетической информацией и биологическими фенотипами. Нежелательные клинические результаты могут возникнуть из-за неопределенности, связанной с модификацией генома в сложных биологических системах, что создает дилеммы при обсуждении этических вопросов [123,124]. Поэтому необходимо всестороннее обсуждение общественных последствий с участием ученых, законодателей и специалистов по этике, поскольку использование и патентование методов генотерапии в лечебных целях вызвало раскол в научных кругах [123]. Однако следует отметить, что каждый из этих текущих и перспективных достижений в разработке и совершенствовании систем редактирования генома на основе CRISPR/Cas9, несомненно, произведет революцию в области медицины в ближайшее десятилетие и, возможно, обеспечит окончательное излечение от β-гемоглобинопатий.
6. Conclusions
CRISPR/Cas9 проложила путь к новым терапевтическим вмешательствам в генетические заболевания человека, таким образом, оказавшись перспективной стратегией лечения наследственных заболеваний крови. Традиционные методы лечения, такие как переливание крови, хелатирование железа и фармацевтические препараты, могут временно уменьшить тяжесть клинических проявлений β-талассемии, но не обеспечивают устойчивого лечения этого заболевания. В настоящее время HSCT и генотерапия на основе вирусных векторов являются единственными вариантами лечения, которые предлагают потенциальное постоянное излечение ТДТ. В то время как отсутствие гистосовместимости донора снижает шансы на использование HSCT в качестве лечебной стратегии, вирусы, используемые в генотерапии, могут повышать риск развития рака, что ограничивает использование этих подходов в качестве лечебной терапии TDT. Невирусные методы генотерапии, такие как липосомальная доставка генов, полимерная доставка генов и микроинъекция ДНК, кажутся привлекательными альтернативами вирусным векторам благодаря их преимуществам в плане безопасности, включая сниженную патогенность и простое недорогое производство. Тем не менее, низкая эффективность трансфекции остается критическим препятствием для клинического применения невирусных методов доставки генов. Таким образом, в настоящее время акцент смещен в сторону генотерапии CRISPR/Cas9 как перспективного терапевтического инструмента для лечения β-гемоглобинопатий. Было показано, что терапия CTX001, основанная на редактировании генов CRISPR/Cas9 ex vivo, улучшает уровень HbF в аутологичных CD34+ HSPC, отредактированных CRISPR, и может снизить потребность в переливании крови у пациентов с TDT на протяжении всей жизни. Помимо юридических и этических соображений и серьезной проблемы внецелевого мутагенеза, генотерапия ex vivo, опосредованная CRISPR/Cas9, подает большие надежды как многообещающая одноразовая терапевтическая стратегия для лечения β-талассемии.
|