Посещений: 
РЕДАКТИРОВАНИЕ ДНК и РНК
CRISPR-Cas инструменты
Assessing and advancing the safety of CRISPR-Cas tools: from DNA to RNA editing • Jianli Tao,
• Daniel E. Bauer &
• Roberto Chiarle
 Nature Communications volume 14, Article number: 212 (2023)
|
CRISPR-Cas gene editing has revolutionized experimental molecular biology over the past decade and holds great promise for the treatment of human genetic diseases. Here we review the development of CRISPR-Cas9/Cas12/Cas13 nucleases, DNA base editors, prime editors, and RNA base editors, focusing on the assessment and improvement of their editing precision and safety, pushing the limit of editing specificity and efficiency. We summarize the capabilities and limitations of each CRISPR tool from DNA editing to RNA editing, and highlight the opportunities for future improvements and applications in basic research, as well as the therapeutic and clinical considerations for their use in patients.
|
Редактирование генома, включающее точное и эффективное манипулирование последовательностями ДНК в геноме человека для изменения судьбы клеток и признаков организма, обладает потенциалом лечения генетических заболеваний1,2. В 2012 году бактериальный кластерный регулярно перемежающийся короткими палиндромными повторами (CRISPR)-CRISPR-ассоциированный белок 9 (Cas9) был сконструирован для РНК-программируемого разрезания ДНК3. С тех пор системы CRISPR-Cas использовались для манипулирования геномами культивируемых и первичных клеток4-7, животных8 и растений9, способствуя значительным достижениям в области наук о жизни и проходя многочисленные клинические испытания10-12. В свете растущего числа новых типов CRISPR-Cas13 , в данном обзоре мы сосредоточились на CRISPR-Cas9/Cas12 , поскольку они являются наиболее широко используемыми редакторами генома.
CRISPR-Cas редактирование ДНК вызывает двунитевые разрывы (DSBs), которые восстанавливаются преимущественно путем негомологичного соединения концов (NHEJ) и гомологией направленной репарации (HDR). В то время как NHEJ приводит к небольшим вставкам или делециям (indels) и нарушению работы генов в целевых сайтах мишенях, гомологически-направленная репарация (HDR) может привести к встраиванию последовательности из экзогенного шаблона ДНК в сайт DSB, хотя эффективность может быть ограничена и неактивна в покоящихся клетках14. В отличие от этого, редакторы оснований и прайм-редакторы точно устанавливают целевые точечные мутации, не требуя DSBs или шаблонов донорской ДНК15.
CRISPR-ассоциированные транспозазы, Tn6677 и CAST, позволяют интегрировать более крупные ДНК-грузы, но до сих пор проявляли активность только в бактериальных клетках16,17. Вкратце, эта система использует комбинацию из нескольких компонентов для внесения желаемых грузов (до 10 kb) в целевые локусы, включая оперон транспозазы (TnsA, TnsB, TnsC, TniQ), субстрат ДНК для донорской транспозазы - (LE-cargo-RE) и ДНК-связывающие и генерирующие R-петли белки на основе CRISPR (слитые гены cas6, cas7 и cas8-cas5 и соответствующий массив CRISPR для Tn6677, или cas12k и массив направляющей (guide) РНК для CAST). Инженерные интегразы/рекомбиназы, слитые с Cas9, которые теоретически могут вставлять, удалять, инвертировать или заменять целевую ДНК, также были использованы для изменения геномной ДНК в клетках млекопитающих, хотя на сегодняшний день с низкой эффективностью и существенными ограничениями последовательности мишени18,19.
В дополнение к редактированию ДНК, аденозиндезаминазы, действующие на РНК (ADAR), слитые с dCas13, были использованы для достижения точного аденозин -> инозинового редактирования целевой РНК20. В отличие от редактирования генома, которое является постоянным, эффекты редактирования РНК преходящи и обратимы. Точное редактирование РНК также может быть достигнуто путем рекрутирования эндогенных ADARs с помощью разработанных ADAR-recruiting RNAs, без необходимости введения чужеродных ферментов редактирования или экзогенного белка ADARs в редактируемые клетки21,22. Таким образом, редактирование оснований РНК имеет терапевтический потенциал как дополнительный подход к редактированию генома.
В этом обзоре мы рассказываем о последних достижениях в разработке и применении инструментов редактирования генома и РНК на основе CRISPR, включая нуклеазы CRISPR-Cas9/Cas12/Cas13, редакторы оснований ДНК, редакторы праймеров и редакторы оснований РНК, уделяя особое внимание повышению эффективности и специфичности их редактирования. Мы также кратко описываем различные методы на основе NGS, которые были разработаны для изучения непреднамеренных генетических изменений и оценки проблем безопасности при редактировании генов. Наконец, мы представляем перспективы будущих направлений, а также терапевтические и клинические соображения в этой быстро развивающейся и захватывающей области.
CRISPR-Cas9/Cas12 nucleases
Системы CRISPR-Cas второго класса были классифицированы на три типа - тип II, тип V и тип VI, с соответствующими эффекторными белками Cas9, Cas12 и Cas13, соответственно. Среди них большинство вариантов Cas9 и Cas12 обладают эндонуклеазной активностью в отношении ДНК, а варианты Cas13 склонны преимущественно к РНК-мишеням и обладают расщепляющей активностью15.
Нуклеазы Cas9 направляются одиночными направляющими РНК (sgRNAs), которые были созданы путем слияния CRISPR РНК (crRNA) и транс-активирующей crRNA (tracrRNA) в одну молекулу РНК, преимущественно создавая тупо-заканчивающиеся концы при двухцепочечном разрыве (DSB) в последовательности протоспейсера 3. Механистически, после образования R-петли рибонуклеопротеидным комплексом Cas9 и направляющей РНК, нуклеазный домен HNH Cas9 расщепляет связанную с направляющей РНК целевую нить ДНК мишени, а RuvC-подобный нуклеазный домен разрезает PAM-содержащую нецелевую нить ДНК на 3 п.н. выше PAM 23,24 (рис. 1a). Cas9 также может создавать ступенчатые разрезы, которые приводят к предсказуемым вставкам длиной в 1 п.н. 25-27. В отличие от Cas9, многие нуклеазы Cas12 естественным образом направляются одной crRNA. Более того, нуклеазы Cas12 обладают только одним RuvC-подобным нуклеазным доменом, который обеспечивает расщепление обеих нитей ДНК-мишени, генерируя ступенчато-образные DSB-разрезы в областях протоспейсера, которые находятся дистальнее последовательности PAM 28 (рис. 1б).
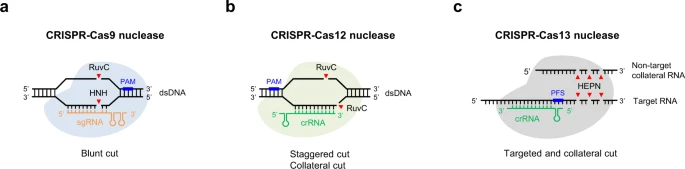 Fig. 1: Overview of CRISPR-Cas nucleases.
a-c CRISPR-Cas9 nucleases and CRISPR-Cas12 nucleases are RNA-guided DNA editing tools, whereas CRISPR-Cas13 nucleases have RNA-guided RNA endonuclease activity. Besides targeted cutting, CRISPR-Cas12/13 nucleases also have collateral cutting activity on non-target DNA/RNA templates. Streptococcus pyogenes (SpCas9) recognizes a relatively common 3' NGG PAM, and functions optimally with 20-nt spacers, while Cas12a orthologs generally use 5' T-rich PAMs. PAM protospacer adjacent motif, PFS protospacer flanking site. Genome-wide off-target editing
Хотя специфичность целевой ДНК нуклеазы CRISPR-Cas определяется направляющей РНК, белки Cas могут связывать и расщеплять частично комплементарные вне-целевые последовательности посредством ряда неканонических взаимодействий сопряжения оснований внутри гетеродуплекса из направляющей и вне-целевой последовательность 29,30 , это вызывает опасения по поводу безопасности их использования в клинических приложениях и настоятельную необходимость систематического выявления этих внецелевых событий. Вкратце, методы геномной идентификации и характеристики потенциальных сайтов вне-целевого действия нуклеазы CRISPR-Cas делятся на три общие категории: биоинформационное предсказание in silico на основе гомологии последовательностей, идентификация сайтов расщепления in vitro с использованием шаблонов геномной ДНК и захват in cellulo событий вне-целевого редактирования 31 (Таблица 1). Однако ни один инструмент не может точно предсказать все события вне-целевого редактирования, особенно те, которые происходят с низкой частотой. Кроме того, предполагаемые вне-целевые сайты требуют дальнейшего подтверждения с помощью направленного глубокого секвенирования, которое является золотым стандартом анализа для проверки наличия индуцированных нуклеазой indels и мутаций 32. Помимо направленного глубокого секвенирования, другие методы проверки, такие как CUT-PCR33 и TIDE/TIDER34 , сильно ограничены масштабами проверки, а также ограничены чувствительностью обнаружения в случае TIDE/TIDER 34 на основе sanger-секвенирования. Тем не менее, интеграция библиотек сайтов-кандидатов вне мишени с помощью SURRO-seq 35,36 и улучшенная система двойной мишени 37 позволяют проводить широкомасштабную оценку действия вне мишеней нуклеазы CRISPR-Cas, выявленных другими методами, в геномном и клеточном контексте, хотя эти методы все еще не могут полностью имитировать доставку редактора и состояние хроматина, ожидаемое в клетках-мишенях для терапии редактирования генов.
Table 1 A summary of methods for identifying genome-wide CRISPR-Cas off-target sites
Cas-OFFinder - это широко используемое in silico программное обеспечение для предсказания Cas9-индуцированных эффектов вне мишеней38. Для ранжирования потенциальных эффектов вне мишеней для определенных направляющих РНК в геноме было разработано несколько алгоритмов прогнозирования на базе Интернета, включая CasOT, E-CRISP, COSMID, CROP-IT, CCTOP, Bowtie2, CNN_std, Elevation, FlashFry, predictCRISPR, Crisflash, Synergizing CRISPR, CRISPRitz, MOFF и многие другие, которые были недавно обобщены31,32 (Таблица 1). Вычислительные алгоритмы, основанные на сходстве последовательностей, хотя и являются мощными и полезными, могут нуждаться в большем количестве обучающих данных и более богатой биофизической базе для полного прогнозирования вне-целевого потенциала. В настоящее время прогнозы in silico должны быть дополнены экспериментальными методами для эффективного определения истинных сайтов эффектов вне мишеней.
Одним из классов таких методов, основанных на экспериментах, является определение off-target сайтов in vitro с использованием очищенной геномной ДНК и рибонуклеопротеинового (RNP) комплекса Cas9 или Cas12 gRNA. Digenome-seq - это широко используемый метод, основанный на переваривании in vitro Cas нуклеазой цельного генома (WGS) 39. Такое переваривание in vitro дает последовательность чтений с одинаковыми 5' концами в местах расщепления, которые могут быть идентифицированы вычислительным путем 39 (рис. 2). Для имитации хроматиновой среды в эукариотических клетках на основе Digenome-seq был разработан DIG-seq, который использует нативный хроматин для переваривания in vitro в отличие от очищенной геномной ДНК в Digenome-seq и, таким образом, увеличивает скорость оценки 40. Основанные на WGS Digenome-seq и DIG-seq сильно ограничены своей стоимостью, поскольку они требуют высокого покрытия секвенирования (~400-500 миллионов считываний), и почти все чтения секвенирования являются фоновыми считываниями (не связанными с CRISPR-Cas индуцированными DSBs). CIRCLE-seq использует альтернативный геномный субстрат, циркулярную ДНК, для геномной идентификации нуклеазных мишеней in vitro41. Механистически, геномная ДНК разрезается на линейные фрагменты, превращенные в циркулярные путем внутримолекулярного лигирования и инкубируется с нуклеазой Cas, во время которого закольцованные молекулы ДНК, содержащие сайт вне мишени, линеаризуются. Эти линеаризованные фрагменты затем селективно амплифицируются и секвенируются для выявления внецелевых участков 41 (рис. 2). CIRCLE-seq значительно снижает фон эндогенных DSBs, возникающих в ходе биологических процессов, или случайных DSBs, возникающих при очистке и обработке геномной ДНК, а также уменьшает глубину секвенирования (~4-5 миллионов считываний), что делает этот метод высокочувствительным. Кроме того, CIRCLE-seq был усовершенствован CHANGE-seq, масштабируемым, автоматизируемым методом на основе тегирования для измерения геномной активности нуклеазы Cas in vitro42. CIRCLE-seq и CHANGE-seq ограничены проблемами, связанными с созданием большого количества высококачественного материала, что делает их гораздо более специализированными, чем Digenome-seq или DIG-seq. Вместо секвенирования всех фрагментов ДНК в Digenome-seq, был разработан SITE-seq для обогащения фрагментов, переваренных нуклеазой, путем селективного мечения расщепленной ДНК биотин-меченым адаптером с последующей аффинной очисткой биотин-стрептавидином и секвенированием с минимальной глубиной чтения (~0,62-2,46 млн. считываний) 43 (рис. 2). Как и другие методы in vitro, вне-целевые мишени, обнаруженные с помощью SITE-seq, нуждаются в дальнейшем подтверждении с помощью методов in cellulo.
 Fig. 2: Methods for identifying genome-wide CRISPR-Cas off-target sites.
Genome-wide methods for off-target detection can be divided into two groups: cell-free methods (in vitro) and cell-based methods (in cellulo). Cell-free methods include Digenome-seq (digested genome sequencing) and its improved version DIG-seq that use genomic DNA or chromatin as template during in vitro digestion; CIRCLE-seq (circularization for in vitro reporting of cleavage effects by sequencing) and its improved version CHANGE-seq (circularization for high-throughput analysis of nuclease genome-wide effects by sequencing) that use DNA circles as template; and SITE-seq (selective enrichment and identification of tagged genomic DNA ends by sequencing) that tags the DSBs with biotin-labeled adapter for enrichment. CROss-seq (CRISPR Off-targeting ssDNA sequencing) could be used both in vitro and in cellulo, capturing CRISPR-Cas induced R-loops by N3-Kethoxal labeling. Cell-based methods include GUIDE-seq (genome-wide, unbiased identification of DSBs evaluated by sequencing) and its improved versions iGUIDE and GUIDE-tag that utilize dsODNs insertions at DSBs in cellulo; HTGTS (high-throughput, genome-wide translocation sequencing), as well as its improved versions LAM-HTGTS and PEM-seq, and similar methods CAST-seq and UDiTaS, can capture translocations between CRISPR-Cas induced on-target (as "bait") and off-targets (as "preys"); IDLV capture (integrase-deficient lentiviral vector) uses lentiviral vector integrations to identify off-targets, while a similar technique ITR-Seq identifies off-targets by capturing the insertions of specific AAV vector sequences called inverted terminal repeats (ITRs); PolyA-seq detects off-targets by capturing de novo LINE-1 retrotransposon insertions at CRISPR-Cas induced DSBs; PEAC-seq (Prime Editor Assisted off-target Characterization), as well as a similar technique TAPE-seq (TAgmentation of Prime Editor sequencing), tags the on- and off-targets by adopting the prime editor with Cas9 nuclease and a sequence-optimized tag-containing pegRNA; DISCOVER-Seq (discovery of in situ Cas off-targets and verification by sequencing) tracks the precise recruitment of MRE11 at DSBs; and BLESS (direct in situ breaks labeling, enrichment on streptavidin, and next-generation sequencing), as well as its improved version BLISS (breaks labeling in situ and sequencing), directly captures unrepaired DSBs by in situ ligation of biotinylated adapters (for BLESS) or T7 promotor sequence-containing adapters (for BLISS) into off-target breaks. Хотя бесклеточные биохимические методы in vitro являются быстрыми и традиционными, они неизбежно захватывают некоторые псевдо вне мишени сайты по сравнению с методами in cellulo, которые требуют введения нуклеазы Cas непосредственно в клетки для выявления сайтов вне мишени в масштабах всего генома. Эти несмещенные методы, основанные на NGS, делятся на четыре категории: косвенное маркирование внецелевых разрывов продуктами репарации (GUIDE-seq, HTGTS, IDLV capture, PolyA-seq, PEAC-seq), косвенное маркирование внецелевых разрывов белками репарации ДНК (DISCOVER-seq), прямое маркирование не-репарированных концов разрывов in situ (BLESS, BLISS) и косвенное маркирование внецелевых сайтов путем захвата R-петли, опосредованной ssDNA (CROss-seq) (Таблица 1).
GUIDE-seq является наиболее используемым методом, который заключается во вставке линейных двухцепочечных олигодезоксинуклеотидов (dsODNs) в сайты CRISPR-Cas вне мишени44. После вставки dsODNs специфическая последовательность и соседние геномные последовательности селективно амплифицируются и секвенируются (рис. 2). Используя более крупные dsODNs (46 нт против 34 нт), GUIDE-seq был улучшен с помощью iGUIDE, что позволяет отличить артефакты неправильного прайминга от подлинных сайтов интеграции dsODNs45. Кроме того, GUIDE-tag использует связывание между нуклеазой Cas9 (SpyCas9-mSA) и донорской ДНК (биотин-iGUIDE донор) для увеличения скорости захвата вне мишени, а также включение UMI через метку Tn5 для предотвращения смещения PCR46. Ограничения методов, основанных на GUIDE-seq, включают требование высокой эффективности трансфекции dsODNs в клетки и ограниченное использование для DSBs с тупыми концами. HTGTS был первоначально разработан как мощный метод, который может генерировать геномную карту хромосомных транслокаций из фиксированной DSB "приманки", индуцированной либо I-SceI47 , либо CRISPR-Cas948. HTGTS использует хромосомные транслокации между индуцированными нуклеазой Cas целевыми DSB ("приманка") и вне-целевыми DSB ("добыча") для захвата вне-целевых сайтов в масштабах всего генома (рис. 2). LAM-HTGTS49,50 повышает чувствительность при меньшей стоимости, а PEM-seq51,52, а также две аналогичные методики CAST-seq53 и UDiTaS54, получили дальнейшее развитие для систематического и количественного расчета частоты различных результатов редактирования (т.е. indels, вне мишеней и хромосомных структурных изменений), что особенно полезно для доклинических и клинических оценок. Хотя методы на основе HTGTS являются мощными и обладают высокой чувствительностью, ограничение заключается в том, что Cas индуцированные нуклеазой транслокации по своей природе являются редкими событиями, которые требуют большого количества входных геномов для эффективного обнаружения. IDLV capture55 и ITR-Seq56 используют дефектный по интегразе лентивирусный вектор и адено-ассоциированный вирусный (AAV) вектор для выявления эффектов вне мишеней, соответственно; а PolyA-seq выявляет эффекты вне мишени путем захвата de novo вставок ретротранспозона LINE-1 в CRISPR-Cas индуцированных DSBs57 (рис. 2). Чувствительность IDLV capture, ITR-Seq и PolyA-seq ниже, чем у других методов in cellulo, из-за того, что эти методы сохраняют высокую способность к случайной интеграции в геном; другими словами, большинство интеграций, обнаруженных при IDLV capture, ITR-Seq и PolyA-seq, являются геномными вставками вне DSBs, индуцированных Cas-нуклеазой. Как и GUIDE-seq, PolyA-seq в значительной степени зависит от эффективной доставки плазмиды LINE-1 (~18 kb) в клетки путем трансфекции, а также от эффективной активности ретротранспозиции в клетках. Недавно сообщалось, что PEAC-seq58 и TAPE-seq59 используют прайм-редактор для вставки оптимизированной по последовательности метки в сайты редактирования на мишени и вне мишени, с последующим обогащением меченых областей праймерами для высокопроизводительного секвенирования (рис. 2). Поскольку вставка метки зависит от последовательности sgRNA, PEAC-seq и TAPE-seq обнаруживают вне-целевые участки с высокой специфичностью, в основном исключая маркировку эндогенных DSBs. Хотя эти методы в значительной степени зависят от эффективности прайм-редактирования, которая может различаться для разных pegRNAs и в разных локусах, PEAC-seq успешно применяется для исследований in vivo в мышиных эмбрионах58.
DISCOVER-seq обогащает DSBs путем иммунопреципитации MRE11, белка, который специфически связывается с DSBs в клетках, чтобы непредвзято обнаружить вне-целевые мишени CRISPR in vivo60 (рис. 2). Поскольку MRE11 связывается с несоединенными концами DSB во время редактирования генома, DISCOVER-seq имеет узкое временное окно для захвата индуцированных нуклеазой Cas мишеней на и вне мишеней. Соответственно, идеальный момент времени для проведения анализа DISCOVER-seq должен быть определен экспериментально и может отличаться для разных типов клеток и методов доставки CRISPR-Cas9.
BLESS непосредственно фиксирует невосстановленные DSBs в масштабах всего генома путем лигирования in situ биотинилированных адаптеров на внецелевых разрывах с последующим обогащением биотин-стрептавидином и секвенированием61 (рис. 2). Поскольку BLESS требует относительно большого количества ДНК, что приводит к высокому фону, BLISS был разработан для обнаружения DSBs с низким требованием к количеству ДНК путем линейной амплификации dsODNs (содержащих последовательность промотора T7), меченных адаптерами DSBs через T7-опосредованную транскрипцию in vitro62 (рис. 2). В отличие от других методов, использующих косвенное маркирование внецелевых разрывов продуктами репарации, BLESS и BLISS могут захватывать DSBs, присутствующие только в определенный временной интервал, и больше не могут захватывать DSBs после их репарации. Более того, BLESS и BLISS имеют относительно высокий фон, который может быть вызван ложными DSBs, вносимыми во время фиксации и обработки.
Поскольку структура R-петли образуется во время распознавания сайта, опосредованного РНК CRISPR, противоположная ssDNA открывается, которая может быть помечена N3-кетоксалом63 и подвержена CROss-seq64, представляя собой новый метод идентификации CRISPR вне мишени, основанный на захвате ssDNA как in vitro, так и in cellulo (рис. 2). Основное ограничение заключается в том, что CROss-seq имеет узкое временное окно, поскольку он может захватывать только R-Loop-ориентированные CRISPR-мишени, в значительной степени исключая целевые сайты, которые были расщеплены и/или репарированы с помощью indels. Более того, корреляция между идентифицированными вне-целевыми участками на основе R-петли и истинными вне-целевыми участками, подтвержденными последовательностью ДНК, еще не определена. Тем не менее, другие чувствительные методы, разработанные для обнаружения геномных DSBs, включая γH2AX ChIP-seq65, TC-seq66, DSB-Seq67, END-seq68, DSBCapture69, I-BLESS70, SAR-seq71 и INDUCE-seq72, также могут быть модифицированы и использованы для обнаружения внецелевых сайтов CRISPR.
В настоящее время не существует единого метода, оптимизированного для всех контекстов, в которых необходимо оценить вне-целевую активность нуклеазы CRISPR-Cas. Таким образом, наилучшей стратегией может быть использование нескольких подходов для оценки и подтверждения внецелевых эффектов. Выбор может быть обусловлен рассмотрением сильных и слабых сторон каждого подхода, как указано в таблице 1, а также различными экспериментальными условиями или клиническими требованиями. Например, если клетки могут быть эффективно трансфицированы, GUIDE-seq и PolyA-seq обеспечивают эффективные, чувствительные и довольно простые методы обнаружения внецелевых эффектов. Захват IDLV или ITR-Seq могут стать хорошей альтернативой в случаях, когда вирусная трансдукция более практична и эффективна, чем трансфекция плазмид dsODNs или LINE-1. Методы in vitro могут быть полезны при неэффективности трансфекции и/или трансдукции клеток, хотя выявленные вне-целевые мишени в конечном итоге должны быть дополнительно подтверждены в клеточном контексте. DIG-Seq - это улучшенный метод in vitro, который сохраняет архитектуру хроматина и, следовательно, имеет более высокий уровень надежности по сравнению с Digenome-seq, CIRCLE-seq и SITE-Seq40. Методы на основе HTGTS, как и другие методы захвата DSBs, являются мощными в выявлении вне-целевой активности CRISPR-Cas, которая приводит к образованию DSBs. В отличие от этого, эти методы сложны для событий захвата вне мишени, опосредованных мутациями, таких как некоторые события вне мишени, индуцированные редакторами оснований.
Genomic rearrangements
Хотя редактирование генома с помощью CRISPR-Cas9 открывает большие перспективы для лечения генетических заболеваний человека, оно приводит к смеси намеренных и ненамеренных генетических изменений. Реже DSBs, индуцированные CRISPR-Cas9, могут привести к большим геномным перестройкам, включая крупные хромосомные делеции, инверсии или транслокации25,49,73-81 или даже к более катастрофическим событиям, таким как хромотрипсис82, анеуплоидия83,84, потеря хромосом85=87 и активация p53, которая может обогатить онкогенные клетки88-90. Дополнительные результаты редактирования CRISPR-Cas9 включают интеграцию экзогенных последовательностей, включая лентивирус55,57,91, адено-ассоциированный вирус (AAV)56,92, плазмиды25,57,78,80,93,94 и небольшие фрагменты ДНК44,95. Недавно мы сообщили, что ретротранспозоны LINE-1 могут возникать в местах редактирования CRISPR-Cas9, причем в зависимости от активности обратной транскриптазы (RT) и независимо от консенсусного мотива эндонуклеазы (EN)57. Эти события используют доступность частых DSBs, создаваемых нуклеазой CRISPR-Cas9, для вставки экзогенных фрагментов ДНК с помощью микрогомологического соединения концов (MMEJ)25,57,92,96. Хотя хромосомные перестройки, скорее всего, являются редкими событиями, опасения вызывает тот факт, что даже очень небольшое количество клеток, отредактированных CRISPR-Cas9, несущих эти вредные события, может клонироваться in vivo97 и вызывать заболевания.
Methods for reducing off-target effects and genomic rearrangements
Способ снижения вне-целевых эффектов заключается в оптимизации различных компонентов систем CRISPR-Cas. Выбор sgRNAs, обеспечивающих высокую эффективность редактирования при низкой вне-целевой активности, вероятно, является предпочтительной стратегией для повышения безопасности, особенно для клинического применения. Кроме того, было разработано несколько модификаций для уменьшения вызываемых нуклеазой CRISPR вне-целевых эффектов и геномных перестроек при сохранении надежной целевой активности, включая модификации sgRNAs, оптимизацию белка Cas9, модификации системы Cas9 и усовершенствование доставки.
Усеченные sgRNAs (tru-gRNAs) (содержащие только 17 или 18 нуклеотидов)98, химически модифицированные sgRNAs (2'-O-метил-3'-фосфоноацетат, или модификация "MP")99, деградирующие под действием света sgRNAs100, и встроенные соединенные мостиками нуклеиновые кислоты (2',4'-BNANC[N-Me]) или заблокированные нуклеиновые кислоты (LNA) в определенных местах в crRNAs101 , как было показано, широко снижают вне-целевую активность CRISPR-Cas9 на несколько порядков. Интересно, что поскольку короткие gRNAs (длиной менее 16 нт) эффективно привлекают Cas9 к комплементарным участкам генома, но не позволяют Cas9 проявлять нуклеазную активность, CRISPR GUARD был разработан как метод защиты внецелевых участков путем совместной доставки коротких gRNAs, конкурирующих с целевой sgRNA102, аналогично совместному введению dead-RNAs (dRNAs)103.
Помимо модификаций sgRNA, генерируются и далее оптимизируются высокоточные белки SpCas9. eSpCas9 (K810A или K848A, K1003A и R1060A)104, FeCas9 (K848A, K1003A, R1060A и D1135E)51, и SpCas9-HF1 (N497A, R661A, Q695A и Q926A)105 ослабляют контакты с фосфодиэфирной основой, в результате чего они оказываются в неактивном состоянии при связывании с несовпадающими мишенями, в то время как HypaCas9 (N692A, M694A, Q695A и H698A)106 повышают жесткость аллостерической регуляции активности домена HNH с высокой геномной специфичностью. HiFi Cas9 (R691A) демонстрирует превосходное целевое редактирование с доставкой RNP по сравнению с eSpCas9 и SpCas9-HF1 при сниженной вне-целевой активности107. Кроме того, сообщается, что evoCas9 (M495V, Y515N, K526E и R661Q)108, Sniper Cas9 (F539S, M763I и K890N)109, xCas9 (A262T, R324L, S409I, E480K, E543D, M694I и E1219V)110, Cas9_R63A/Q768A111 и SuperFi-Cas930 обладают более низкой вне-целевой активностью.
Никаза SpCas9 расщепляет только одну нить ДНК путем мутации одного из нуклеазных доменов (D10A или H840A). Двойной никинг парными никазами Cas9 показал существенно повышенную специфичность редактирования генома49,112-115. В лаборатории Wolfe была разработана система Cas9-pDBD (программируемый ДНК-связывающий домен), которая ограничивает его способность задействовать целевой сайт, повышая специфичность до 160 раз на внецелевых сайтах116. Присоединение минимального мотива к spCas9, MiCas9, увеличивает частоту нокаута крупных генов и снижает количество нежелательных indels в мишени и вне мишени за счет обогащения RAD51 в целевых сайтах117. Показано, что частота мелких indels и крупных делеций снижается в клетках, дефицитных по гену центральной резекции Nbn и гену микрогомологии, опосредованной концевым соединением Polq81. Лаборатория Hu сообщила, что генерация высокого уровня транслокаций зависит от повторного разрезания в сайтах-мишенях Cas9118. Таким образом, Cas9TX образуется путем слияния Cas9 с оптимизированной TREX2, экзо-эндонуклеазой, которая предотвращает идеальную репарацию ДНК и тем самым избегает повторного расщепления, устраняя хромосомные транслокации при редактировании генома как в клеточных моделях118, так и в мышиной модели возрастной макулярной дегенерации119. Недавно было показано, что по сравнению с нуклеазами Cas9 и Cas12a, нуклеазы Cas12f снижают уровень внецелевых эффектов, хромосомных транслокаций, крупных делеций и плазмидных инсерций80.
CRISPR-Cas могут быть доставлены в клетки либо в виде плазмидной ДНК или мРНК, кодирующей их экспрессию, либо непосредственно в виде белков или рибонуклеопротеинов (RNPs)2. Было показано, что RNPs уменьшают вне-целевые эффекты, поскольку они действуют на целевую ДНК сразу после трансфекции и быстро разрушаются протеазами и рибонуклеазами в клетках120-122. Например, опосредованная экзосомами доставка РНК Cas9 была показана для тканеспецифической генотерапии заболеваний печени123. Более того, учитывая многочисленные интеграции плазмидной ДНК или вирусной ДНК в местах редактирования, RNPs могут стать более безопасным методом доставки инструментов CRISPR-Cas. Хотя эндогенные ретротранспозоны LINE-1 все еще могут представлять потенциальную угрозу при редактировании CRISPR-Cas с помощью RNPs, редакторы оснований или прайм-редакторы могут быть использованы в качестве более безопасной альтернативы, поскольку они уменьшают количество DSBs57.
DNA base editors
Многие приложения по редактированию генома требуют введения точных точечных мутаций, небольших вставок или делеций для установки или исправления патогенных мутаций в геноме человека 15. Два класса редакторов оснований ДНК, редакторы цитозиновых оснований (CBE) и редакторы адениновых оснований (ABE), были разработаны для точной индукции целевых точечных мутаций, не требующих DSBs или шаблонов донорской ДНК 14. В CBEs цитидиндеаминаза APOBEC/AID сливается с никазой Cas9 для целенаправленного воздействия через sgRNA и с UGI для повышения точности и эффективности редактирования оснований, преобразуя пары оснований C•G в пары оснований T•A 124,125. В ABEs деаминаза TadA, созданная в лаборатории, сливается с никазазой Cas9 для преобразования пар оснований A•T в пары оснований G•C 126. Аналогично, редакторы оснований C-to-G (CGBEs) (такие как GBE127, CGBE1128 и другие CGBEs129,130,131) состоят из никазы Cas9, деаминазы цитидина и гликозилазы урацил-ДНК (Ung), преобразуя пары оснований C•G в пары оснований G•C, расширяя область редактирования редакторов оснований (рис. 3a).
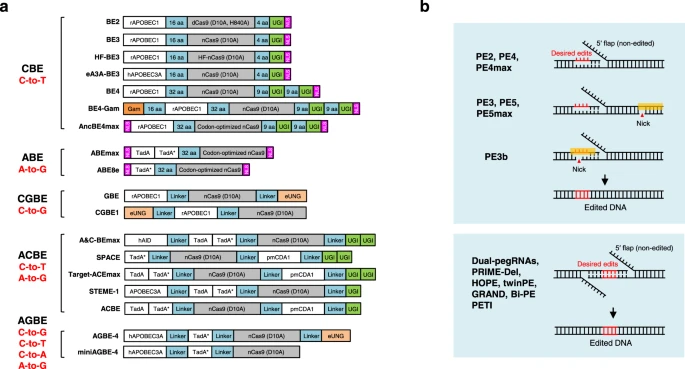 Fig. 3: Overview of DNA base editors and prime editors.
a Diagram of the DNA base editors. BE2, BE3, HF-BE3, eA3A-BE3, BE4, BE4-Gam, and AncBE4max are developed as cytosine base editors (CBEs), while ABEmax and ABE8e are adenine base editors (ABEs). GBE and CGBE1 belongs to C-to-G base editors (CGBEs). A&C-BEmax, SPACE, Target-ACEmax, STEME-1 and ACBE are generated as dual-deaminase base editors (ACBEs) by fusing CBE with ABE. When fused a CGBE with ABE, AGBEs are developed as represented by AGBE-4 and miniAGBE-4. b Diagram of the Prime editors. While nCas9 (H840A) and pegRNA are required for all prime editing strategies, PE3/PE5/PEmax contains a nicking sgRNA to increase the editing efficiency. PE3b contains a sgRNA with spacer that match the edited strand to minimize the presence of concurrent nicks. Besides one pegRNA-mediated conventional PEs, two pegRNAs strategy is used in dual-pegRNAs, PRIME-Del, HOPE, twinPE, GRAND, Bi-PE and PETI to further increase the prime editing efficiency, enable longer edits and introduction of recombination sites.
Вкратце, в отличие от dCas9-слитого редактора BE2, BE3 использует никазазу Cas9 (D10A) для специфического связывания не редактируемой нити для повышения эффективности редактирования124,132. Увеличивая длину линкера и добавляя вторую копию UGI, BE4 осуществляет редактирование оснований с более высокой эффективностью и значительно улучшенной чистотой продукта по сравнению с BE3133. Соединение BE4 с Gam, белком бактериофага Mu, который связывает DSBs, BE4-Gam значительно снижает образование indels во время редактирования оснований133. Оптимизация использования кодонов и последовательности ядерной локализации (NLS) привела к созданию наиболее совершенного и эффективного AncBE4max или ABEmax134, хотя эта повышенная активность может также увеличить вне-целевые эффекты. ABEmax был оптимизирован на основе ABE7.10 и состоит из мономера TadA дикого типа и эволюционировавшего мономера TadA, при этом было показано, что мономер TadA дикого типа не требуется для активности ABE135,136. Таким образом, ABE8e был разработан только с одним фаго-ассоциированным далее эволюционировавшим мономером TadA, показав самую высокую эффективность136 (рис. 3a).
Кроме того, двойные деаминазы оснований (ACBE) (включая A&C-BEmax137, SPACE138, Target-ACEmax139, STEMEs140 и ACBE141) были созданы путем слияния цитидиндеаминазы и адениндеаминазы вместе с никазазой Cas9 для одновременного осуществления преобразования C-в-T и A-в-G на одном и том же сайте-мишени. Совсем недавно в лаборатории Lai объединили CGBE с ABE для создания нового типа системы редактирования оснований, опосредованной двойной деаминазой, названной системой AGBE, которая может одновременно вводить 4 типа преобразований оснований (C-в-G, C-в-T, C-в-A и A-в-G), а также indels и, таким образом, может быть использована для насыщенного мутагенеза142 (рис. 3a).
Off-target DNA editing and RNA editing of DNA base editors
Хотя редакторы оснований ДНК могут быть более точными инструментами по сравнению с нуклеазой CRISPR-Cas57 , чувствительное и объективное обнаружение их внецелевых эффектов остается важным для применения base editors (BE) в терапии человека. Существует два типа внецелевой активности CBEs и ABEs: Cas-зависимые вне-целевые события, вызванные сходством последовательностей в пределах заданной толерантности к несоответствиям, и Cas-независимые вне-целевые события, возникающие в результате нецелевого связывания деаминазы с вне-целевыми участками. С помощью GOTI (геномный анализ внецелевых участков путем инъекции в двухклеточные эмбрионы) лаборатория Yang показала, что в мышиных эмбрионах CBEs генерируют геномные значительные вне-целевые однонуклеотидные варианты, которые обогащены в транскрибируемых областях генома143, подобно другой превосходной работе на рисе144. Эти результаты были подтверждены и расширены с помощью Digenome-seq145,146, EndoV-seq147, Orthogonal R-loop assay148,149 и CROss-seq64, показавших, что ABE также могут генерировать вне-целевые варианты в масштабах всего генома с гораздо меньшей частотой.
Помимо DNA off-targeting, для обнаружения off-таргетинга на уровне РНК также использовался транскриптомное РНК-секвенирование. Три независимые группы показали, что как CBEs, так и ABEs могут вызывать значительные (десятки тысяч) SNVs РНК вне мишени в клетках человека135,150-152, что соответствует способности деаминазы APOBEC1 или TadA редактировать РНК.
Out-of-protospacer editing and target-strand editing of CBE
CBEs катализируют превращения C в dU и в конечном итоге приводят к переходам C в T124. Захват дезоксиуридина (dU) хорошо зарекомендовал себя в таких высокопроизводительных методах NGS, как Excision-seq153, dU-seq154, UPD-Seq155, U-DNA-Seq156 и AI-seq157. Благодаря захвату промежуточного продукта редактирования оснований dU, более продвинутый метод Detect-seq158 способен отслеживать как целевые, так и нецелевые события редактирования CBE, что недавно было подтверждено аналогичным методом под названием Ucaps-seq159. Механистически, Detect-seq использует восстановленную in vitro реакцию репарации эксцизии оснований (BER) для достижения специфического мечения dU, в ходе которой обычные dTTP или dCTP заменяются на биотин-dUTP или 5-формил-дезоксицитидинтрифосфат (5fdCTP), чтобы обеспечить биотиновое вытягивание dU-содержащей ДНК или тандемные переходы C-в-T (используемые для усиления сигнала), соответственно158. Помимо Cas9-зависимого и Cas9-независимого off-targeting, Detect-seq обнаружил CBE-редактирование за пределами последовательности протоспейсера (out-of-protospacer editing) и на не редактируемой нити (target-strand editing), расширяя знания о том, что CBE могут вызывать ближайшие вне мишени мутации158.
DddA-производные митохондриальные редакторы оснований (DdCBEs), которые представляют собой слияние расщепленных половинок DddA и белков массива транскрипционных активаторов-подобных эффекторов (TALE), позволяют осуществлять целенаправленные преобразования C-в-T именно в митохондриальной ДНК160. Интересно, что, используя ту же технику, компания Detect-seq обнаружила, что DdCBEs могут вызывать обширное внецелевое редактирование в ядерном геноме в TALE array sequence (TAS)-зависимой или TAS-независимой манере161. Важно отметить, что авторы сконструировали DdCBEs тремя различными способами для ослабления таких ядерных внецелевых эффектов.
Methods for reducing off-target effects of DNA base editing
Стратегии снижения внецелевых эффектов редактирования оснований ДНК аналогичны нуклеазам CRISPR-Cas, включая модифицированные направляющие РНК, модифицированный Cas9 белок, усовершенствование доставки и использование модифицированной высоко доступной цитидин- или аденозиндезаминазы.
Варианты Cas9 высокой точности, такие как eSpCas9, SpCas9-HF, HypaCas9 и SuperFi-Cas9 в форме никазы, были включены в архитектуру BE для снижения Cas-зависимых внецелевых эффектов132,162,163. Стратегия Cas-embedding, при которой деаминаза (APOBEC1 или TadA) вводится в середину Cas9n в толерантных сайтах вместо N-конца nCas9, значительно снижает независимое от gRNA вне-целевое редактирование редакторами оснований ДНК164. В отличие от доставки плазмидной ДНК/вирусов, доставка BE-систем через комплекс РНК, мРНК/sgRNA или липидные наночастицы (LNPs) приводит к более скоротечной активности редактирования и, следовательно, к улучшению специфичности с меньшим количеством вне-целевых эффектов132,145.
С помощью инженерии цитидин- или аденозиндезаминазы было получено несколько вариантов BE с высокой точности, позволяющих минимизировать эффекты вне мишени в ДНК и РНК165. В CBEs эти варианты включают SECURE-BE3150, eA3A-BE3152,166, hA3G-CBE167, AALN-BE4148, YE1-BE4148, transformer BE (tBE)168, AID-2S/AID-3S169 и другие CBEs нового поколения170,171. В ABEs были созданы SECURE-ABE135, ABE7.10-F148A152, ABEmaxAW или ABEmaxQW151, ABE8e-V106W136, ABE-del153172 и ABE9 (N108Q/L145T)173 , чтобы уменьшить эффекты вне мишени РНК и сузить окно редактирования. Следует отметить, что многие из этих мутаций могут снижать активность деаминазы. Таким образом, повышение специфичности может быть компромиссом за снижение эффективности в селективных терапевтических контекстах.
Поскольку ABE проявляют меньшую активность вне мишени ДНК и РНК, меньший размер и более высокую эффективность редактирования по мишени, чем CBE, две группы реконструировали ABE для эффективного и специфического редактирования оснований цитозина на основе CRISPR174,175. Эволюционировавшие цитидиндеаминазы TadA содержат мутации в ДНК-связывающих остатках, которые изменяют селективность фермента в пользу дезоксицитидина, а не дезоксиаденозина, представляя собой первые неестественные цитозиндеаминазы, помимо естественных белков семейства AID/APOBEC. Вкратце, Td-CGBE осуществляет высокоэффективное и точное редактирование C-в-G175, в то время как Td-CBEs175 и TadCBEs174 генерируют точные преобразования оснований C-в-T со слиянием UGI и дополнительных мутаций. Более того, было показано, что двухбазовый редактор TadA (TadDE) одинаково эффективно редактирует цитозин и аденин одновременно (C-в-T и A-tв-G)174, обеспечивая высокоэффективную и малогабаритную альтернативу, помимо ранее описанных двойных редакторов, которые объединяют цитидин- и аденозиндезаминазы (ACBEs)137-141, а также позволяя проводить насыщающий мутагенез и скрининг.
Prime editors
Праймер редакторы (PE) позволяют точно редактировать геном, включая целевые небольшие вставки, небольшие делеции и все 12 возможных типов точечных мутаций и их комбинаций, не требуя DSBs или шаблонов донорской ДНК. Для PE используется никаза Cas9 (H840A), соединенная с преобразованной вирусом мышиной лейкемии Молони (M-MLV) обратной транскриптазой (RT), которая запрограммирована с желаемой edit-containing pegRNA. PegRNA содержит sgRNA, сайт связывания праймера (PBS) и reverse RNA template (RTT) для обратной транскриптазы, чтобы создавать желаемые правки в сайте мишени 176-178.
В частности, PE2 формирует вырезку (nicks) целевого участка, чтобы обнажить 3'-OH, который может быть использован для запуска обратной транскрипции pegRNA в редактируемую нить, а PE3 делает вторую выемку на не редактируемой нити, чтобы вызвать ее замену и, следовательно, повысить эффективность редактирования. Затем была разработана улучшенная стратегия PE3b, которая делает вырезки (nicks) на не редактируемой нити только после устранения (resolution) заслонки (flap) на редактируемой нити, чтобы минимизировать наличие одновременных зазубрин и потенциальных DSB при PE3176 (рис. 3b). Кроме того, PE4 и PE5 были разработаны на основе систем PE2 и PE3 соответственно, повышая эффективность редактирования за счет совместной экспрессии доминантно-отрицательного MLH1 (MLH1dn) для ингибирования репарации несоответствий ДНК (MMR)179, что согласуется с другой работой180. Оптимизация белков праймер-редактора привела к наиболее продвинутым PE4max и PE5max179, а эффективность редактирования может быть дополнительно увеличена синонимичными мутациями в pegRNA181 или преобразованными pegRNA для стабилизации структуры РНК179,181-185. Другие оптимизации прайм-редактирования включают добавление ДНК-связывающего домена Rad51186, оптимизированного состава NLS187, совместного отбора с пуромицином188, флуоресцентного репортера189,190 или клеточной устойчивости191, слияния пептидов192, переходного ингибирования р53193, расщепленного праймера, несвязанного с обратной транскриптазой185,194 или сплит-интеин праймера187, удаление домена рибонуклеазы H из M-MLV RT и включение вирусного нуклеокапсидного белка с шаперонной активностью нуклеиновой кислоты195. Более того, было показано, что прайм-редакторы на основе нуклеазы Cas9 повышают эффективность PE, но это происходит за счет чистоты продукта188,196,197.
Вместо системы PE, опосредованной одной pegRNA, несколько групп независимо друг от друга заявили, что две pegRNA, кодирующие одинаковые правки в смысловой и антисмысловой нитях ДНК, позволяют осуществлять программируемые замены или большие делеции с более высокой эффективностью редактирования (рис. 3b). В число этих замечательных работ входят двойные pegRNA, разработанные на растениях198, PRIME-Del199, HOPE200, редактирование twin prime (twinPE)201, редактирование GRAND202, Bi-PE203 и PETI204, а также аналогичная техника PEDAR, которая объединяет нуклеазу Cas9 с обратной транскриптазой (PE-Cas9) и комбинирует ее с двумя pegRNA205. Помимо высокоэффективных замен и больших делеций, twinPE позволяет осуществлять точные большие вставки (длина гена более 5000 п.н.) или большие инверсии (40 кб) в клетках человека в сочетании с сайт-специфической сериновой рекомбиназой, что расширяет возможности редактирования генов для исправления больших или сложных патогенных аллелей человека201, аналогично PASTE, в котором используется CRISPR-Cas9 никаза, соединенная с обратной транскриптазой и сериновой интегразой для целевого набора генома и интеграции желаемой полезной нагрузки (до 36 кб)206.
Safety evaluation and improvements of prime editors
Вследствие потенциальных DSBs, создаваемых двумя никами при PE3, наблюдается определенный уровень геномных фрагментов, плазмид и вставок LINE-1 на целевых сайтах, редактируемых PE3, хотя и с гораздо меньшей частотой, чем нуклеазой CRISPR-Cas9; в то время как редактирование PE2 или PE3b имеет редкие вставки, что соответствует их меньшему образованию DSB57,176. Аналогичным образом, использование двух pegRNA также может привести к образованию DSBs; таким образом, необходимо тщательно оценить их вне-целевое и непреднамеренное внутри-целевое воздействие. Кроме того, несколько исследований показали, что обратная транскрипция 3'-удлиненных pegRNA (последовательность RTT-PBS) может происходить на каркасе tracrRNA, что приводит к массивным вставкам последовательности каркаса в сайте редактирования праймера с различной длиной вставки57,176,207. Поэтому разделение pegRNA на sgRNA и отдельную последовательность RTT-PBS (линейную или кольцевую) не только предотвращает деградацию 3'-удлиненных pegRNA под действием нуклеаз185, но и может быть использовано для уменьшения вставок в каркас pegRNA.
Между тем, были исследованы Cas-зависимые и Cas-независимые вне-целевые эффекты прайм-редакторов. Digenome-seq на основе nickase (nDigenome-seq)208, CROss-seq64 и TAPE-seq59 были использованы для скрининга геномных Cas-зависимых внецелевых сайтов систем PE, демонстрируя, что PE обеспечивают высокоспецифичное редактирование генома с очень ограниченным количеством внецелевых эффектов, которые могут быть улучшены высокоточными вариантами Cas9208. С другой стороны, три недавних исследования показали, что PE3 не вызывает значительных Cas-независимых внецелевых мутаций/перестроек в растениях209 или клетках человека210,211 как на геномном, так и на транскриптомном уровне, что свидетельствует о высокой специфичности редактирования его молекул обратной транскриптазы.
RNA editing
Системы класса 2 типа VI с единственным эффекторным белком Cas13 являются единственными известными CRISPR-Cas системами, нацеленными исключительно на РНК, что делает их перспективными в качестве инструментов для нокдауна, обнаружения и редактирования РНК212. Являясь РНК-направляемой РНК-эндонуклеазой (РНКазой), Cas13 состоит из двух доменов HEPN, которые вместе образуют составной активный центр РНКазы, ответственный за каталитическое расщепление РНК после связывания нацеливающей РНК213,214. Cas13a, Cas13b, Cas13d-опосредованная деградация РНК и нокдаун генов демонстрируют высокую эффективность и специфичность по сравнению с РНК-интерференцией, что делает возможным терапевтическое лечение, включая нервные заболевания и патогенные вирусы215-217.
Интересно, что активированные комплексы Cas13-crRNA расщепляют как целевые РНК (цис-расщепление), так и нецелевые сопутствующие РНК (транс-расщепление)218,219 (рис. 1c), это делает возможным чувствительное обнаружение и диагностику нуклеиновых кислот. Например, SHERLOCK (специфическая высокочувствительная ферментативная репортерная разблокировка) была разработана на основе Cas13 для обнаружения нуклеиновых кислот220,221, а также адаптирована для обнаружения SARS-CoV-2 в обновленной версии под названием STOP (SHERLOCK testing in one pot)222. С другой стороны, сопутствующая деградация побочных РНК системой CRISPR-Cas13 ограничила ее применение in vivo. Поэтому были разработаны высокоточные варианты Cas13 с минимальными побочными эффектами, что может еще больше повысить их полезность для направленной деградации РНК в фундаментальных исследованиях и терапевтических попытках223.
RNA base editors
Подобно редакторам оснований ДНК, каталитически инактивированный белок Cas13 (dCas13) был слит с ADAR2 для создания редакторов оснований РНК, таких как REPAIR (RNA Editing for Programmable A to I Replacement)224 и RESCUE (RNA Editing for Specific C-to-U Exchange)225 , которые могут быть использованы для редактирования РНК A-в-I или C-в-U, соответственно. Чтобы свести к минимуму значительное вне-целевое редактирование РНК в REPAIRv1, лаборатория Zhang выделила новый вариант dCas13b-ADAR2DD (E488Q/T375G), названный REPAIRv2, позволяющий осуществлять точное, эффективное и высокоспецифичное редактирование оснований РНК224.
Совсем недавно, путем рекрутирования эндогенных ADAR с помощью различных типов инженерных ADAR-рекрутирующих РНК (arRNAs), было разработано следующее поколение редакторов оснований РНК, а именно RESTORE (рекрутирование эндогенных ADAR к определенным транскриптам для редактирования РНК, опосредованного олигонуклеотидами)21 и LEAPER (использование эндогенных ADAR для программируемого редактирования РНК)22, чтобы обойти проблемы, вызванные эктопической экспрессией ферментов редактирования или белка ADARs. Для повышения эффективности редактирования и снижения побочного вне-целевого редактирования были созданы CLUSTER226, cadRNAs227 и LEAPER 2.0228 как наиболее совершенные редакторы оснований РНК с использованием оптимизированных в силиконе направляющих РНК CLUSTER226 или ковалентно замкнутых циркулярных arRNAs227,228, а также с использованием промотора CMV вместо промотора U6 и обеднением пар уридинов с вне-целевыми аденозинами228.
Therapeutic and clinical considerations
Considerations on therapeutic gene editing in patients
В этом разделе мы рассмотрим факторы, имеющие решающее значение для использования методов редактирования генов для лечения заболеваний человека, включая терапевтические преимущества, эффективность и доставку in vivo, точность и специфичность.
Что касается преимуществ, то в целом они могут быть значительными для генной терапии, которая устраняет основное молекулярное поражение при генетических заболеваниях или направлена на фундаментальные патофизиологические особенности приобретенных заболеваний. Сроки и терапевтическая доля клеток-мишеней для изменения патофизиологии могут варьировать в зависимости от заболевания. При заболеваниях, при которых со временем накапливаются необратимые повреждения органов, проведение генотерапии на ранних стадиях заболевания может быть полезным229. Терапевтическая доля может быть оценена на основании клинических наблюдений при заболеваниях, при которых были описаны результаты соматического мозаицизма или химерной трансплантации230,231; в других случаях этот порог может быть определен только после проведения исследований по терапевтическому редактированию генов. Кроме того, воздействие на подпороговую фракцию клеток может дать промежуточный клинический эффект, а некоторые генетические модификации могут частично облегчить течение болезни, например, путем преобразования нулевого аллеля в гипоморфный232.
Для эффективного редактирования необходимы адекватные системы доставки для достижения достаточного количества генных модификаций (моноаллельных или биаллельных в зависимости от характера заболевания) в необходимой доле клеток. Эффективность зависит не только от внутренней активности генного редактора, но и от способа его доставки в клетки. Для терапии ex vivo - например, нацеленной на кроветворные клетки - доставка часто осуществляется с помощью пульсового воздействия RNP или мРНК/gRNA посредством электропорации120. Для терапии in vivo наиболее распространена доставка с помощью AAV233, хотя также могут применяться наночастицы234 и голые RNPs1. Учитывая, что импульсная доставка генных редакторов в первичные клетки может привести к скромному и короткому воздействию, возможные геномные эффекты, наблюдаемые in vitro, могут переоценить возможные эффекты редактирования. Некоторые непреднамеренные эффекты геномного ремонта, такие как образование микроядер или потеря гетерозиготности82,235, ограничены в зависимости от стадии клеточного цикла, поэтому воздействие на неделящиеся клетки, возможно, позволит избежать этих эффектов (по сравнению с воздействием на делящиеся клетки в культуре in vitro). Другие геномные эффекты могут зависеть от состояния клетки; например, частота эффектов ретротранспозиции может быть связана с клеточной активностью ретротранспозонов57. Следует отметить, что терапия, направленная на мРНК, а не на ДНК, может потребовать длительной доставки или многократного введения.
С эффективностью связана точность, которая перечисляет, сколько модификаций являются идеальными, а сколько - нежелательными. В случае предполагаемой HDR-репарации может иметь место сопутствующая NHEJ-репарация, которая будет произведена в части клеток. Например, при HDR-репарации мутации серповидноклеточной болезни будет происходить несовершенная репарация, которая может преобразовать аллель серповидноклеточной болезни в аллель β-талассемии, что может дать смешанную клиническую картину химерных здоровых, больных серповидноклеточной болезнью и β-талассемией HSCs, каждая из которых производит клетки крови236. Для NHEJ-репарации, направленной на регуляторные элементы генов, длинные делеции могут удалять кодирующие последовательности и приводить к потере функциональности аллелей74; в других случаях внутрикадровые indels могут давать неоморфные эффекты. Аналогичным образом при редактировании оснований сторонние правки или непреднамеренные замены (например, C-в-G в редакторе оснований C-в-T) могут привести к дополнительным фенотипам, например, из-за непреднамеренных аллелей миссенс или нонсенс в дополнение к терапевтическим модификациям.
Специфичность описывает способность редакторов вызывать изменения в других местах генома, помимо предполагаемой мишени. Некоторые из этих внецелевых эффектов могут быть предсказуемы и проверяемы различными методами, как описано выше. Однако другие эффекты могут быть неожиданными, стохастическими или широко распространенными и поэтому могут ускользнуть от внимания целевых анализов. Даже секвенирование всего генома может оказаться нечувствительным анализом, учитывая, что теоретически редкие клональные события могут быть клинически значимыми. По мере уменьшения частоты таких событий (в конечном итоге до единичного события во многих клетках-мишенях), их обнаружение без целевых анализов может стать невозможным. Часто может существовать компромисс между эффективностью редактирования генов, точностью и специфичностью237. Таким образом, короткий импульс экспрессии высокого уровня может обеспечить желаемый баланс эффективности и специфичности121.
Наконец, еще одной проблемой в этой области является определение моделей для измерения эффективности, точности и специфичности. Всестороннее тестирование геномных модификаций в терапевтически значимых контекстах на экспериментальных моделях может быть затруднено, поскольку ожидается, что не-человеческие клетки , возможно, будут иметь другие вне-целевые последовательности по сравнению с клетками человека, учитывая различия в геномной последовательности. Таким образом, эксперименты на не-человеческих моделях могут не полностью имитировать условия in vivo, соответствующие терапевтической доставке. Улучшенные модели, такие как гуманизированные мыши, могут позволить проверить эффективность целевого редактирования и вне-целевые эффекты в определенных тканях (например, в кроветворных клетках), но не в других. Таким образом, тщательное наблюдение и корреляционные молекулярные анализы у субъектов, участвующих в клинических испытаниях, могут быть единственным способом действительно оценить результаты комплексного геномного редактирования. Такие анализы могут быть получены в ходе исследований, сфокусированных на легкодоступных тканях (таких как кроветворные клетки), биопсии ценных тканей, когда их можно собрать, и, возможно, бесклеточной ДНК238 , но они могут быть невыполнимы для других недоступных тканей.
Considerations on clinical risks of gene editing
Поскольку методы редактирования генов все еще находятся на ранней стадии клинической разработки и ни одна терапия на основе CRISPR еще не была одобрена для использования у пациентов239 , их безопасность и связанные с ними клинические риски остаются неопределенными. Хотя все большее количество методов может быть использовано для каталогизации геномных эффектов на мишени и вне ее, соматическая генетическая модификация сама по себе может не предвещать клинических последствий. Клетки спонтанно накапливают повреждения ДНК с течением времени и при делении клеток, поэтому скромное однократное увеличение количества соматических генетических вариаций может быть не особенно заметным. В целом, ожидается, что большинство генетических вариантов будут нейтральными, а немногие варианты, которые могут быть вредными для клеток, не будут иметь клинического значения. Напротив, потенциально наиболее опасной категорией являются варианты, которые вызывают усиление функции, способствующее клональной экспансии и опухолегенности, активируя онкогены или нарушая супрессоры опухоли. До сих пор в клинических исследованиях не было зарегистрировано ни одного примера непреднамеренной опухолегенности терапевтического редактирования генов, что позволяет предположить, что частота таких эффектов крайне низка.
Наконец, клинические соображения должны распространяться на всю процедуру терапевтического редактирования генов, включая любой побочный риск доставки клеток и реакции хозяина, которые могут представлять более вероятные классы клинического риска. Для подходов in vivo это может включать нежелательные реакции хозяина на векторы переноса генов, такие как вирусы (например, AAV, AdV, LV и т.д.) или транспозоны (Sleeping Beauty), которые могут вызывать стрессовые реакции, такие как интерфероновые реакции и реакции повреждения ДНК240,241. Кроме того, известно, что терапия, требующая подготовительного кондиционирования, например, генотоксическая алкилирующая миелоаблативная химиотерапия, которая часто сопровождает трансплантацию гемопоэтических стволовых клеток, или лимфодеплеция, которая сопровождает иммунотерапию, может иметь риски, которые необходимо учитывать в общем профиле риска, но которые не связаны с редактированием генов как таковым. Например, два случая AML наблюдались среди 47 пациентов, прошедших генотерапию серповидно-клеточной болезни242,243. Молекулярные анализы показывают, что инсерционный мутагенез не был причиной этих злокачественных событий, поэтому в узком смысле можно сделать вывод, что генотерапия не была причиной злокачественных новообразований. На самом деле, лентивирусная терапия моногенных заболеваний крови имеет весьма благоприятный профиль безопасности без явлений генотоксичности, наблюдавшихся на протяжении последних лет у более 700 пациентов, несмотря на обязательную доставку полу-случайных геномных вставок в целевые стволовые клетки244. Это наблюдение иллюстрирует, что подсчет геномных модификаций нелегко перевести в количественную оценку клинического риска.
Conclusions and future perspectives
За последние 10 лет произошел значительный прогресс в понимании и манипулировании системами CRISPR-Cas, особенно в области редактирования генома и РНК. Нуклеазы CRISPR-Cas9/Cas12/Cas13, редакторы оснований ДНК, редакторы праймеров и редакторы оснований РНК произвели революцию в науках о жизни, проложив путь к прогрессу в фундаментальных исследованиях и терапевтическом применении. Тем не менее, для создания более точных, эффективных и безопасных инструментов редактирования необходимы дальнейшие работы. Помимо редактирования ДНК и РНК, другие области применения инструментов на основе CRISPR включают регуляцию эпигенома и генов, функциональный скрининг и диагностику, которые были хорошо описаны в недавних обзорах245,246.
Существует множество чувствительных и объективных методов, которые были разработаны для оценки внецелевых проблем во время редактирования генома, расширяя границы специфичности и эффективности редактирования. Несмотря на прогресс, доступные в настоящее время методы обнаружения вне-целевой активности CRISPR-Cas систем все еще неоднородны, не имеют широкого сравнения друг с другом и оптимизированы только для нескольких экспериментальных систем. Кроме того, отсутствует метод, проверенный и одобренный для клинического применения. Поэтому задача состоит в том, чтобы разработать консенсусный метод, который может непредвзято, чувствительно, быстро и экономически эффективно выявлять события CRISPR-Cas вне мишеней в большинстве клеточных систем, включая модели in vivo. Эта потребность еще больше возрастет, когда терапевтическое применение CRISPR-Cas станет более широким. Усовершенствованные методы обнаружения вне-целевой активности будут направлять усилия по разработке высокоточных Cas нуклеаз или цитидин/аденин деаминаз с минимальным Cas-зависимым или Cas-независимым вне-целевым редактированием, проливая свет на будущее направление точной медицины. Следует отметить, что эти минимизированные вне-целевые эффекты сосредоточены не только на уровне ДНК, но и на уровне РНК, особенно для редакторов оснований ДНК, которые могут вызвать значительное вне-целевое редактирование РНК.
В отличие от редактирования ДНК, редактирование РНК предлагает альтернативу редактированию генома в некоторых терапевтических приложениях в обратимой и настраиваемой манере, особенно для редакторов оснований РНК, которым не нужно вводить чужеродную нуклеазу Cas или экзогенный белок ADARs в редактируемые клетки. Избыточная экспрессия белков Cas или ADARs связана с рядом проблем безопасности, включая существенные геномные и/или транскриптомные вне-целевые повреждения, иммуногенность, онкогенность и проблемы с доставкой228. Тем не менее, для улучшения редактирования оснований РНК по-прежнему необходимы будущие работы по специфичности и эффективности, а также более совершенные редакторы оснований РНК или "прайм-редакторы РНК", которые могли бы модифицировать РНК более широким образом для дальнейшего изучения биологии РНК. Кроме того, для специфического и безопасного редактирования РНК необходимо разработать другие типы инструментов редактирования РНК, например, недавно представленный RESTART247, который опирается на направляющую (guide) snoRNA для восстановления индуцированной PTC терминации трансляции. С другой стороны, "эндогенные редакторы оснований или праймеров ДНК", не требующие использования чужеродных Cas-нуклеаз или экзогенных деаминазных белков, стали бы захватывающим прорывом для редактирования генома следующего поколения.
Наконец, для того, чтобы любое из этих соображений было актуальным, терапия должна быть доступна для пациентов. Учитывая, что многие пациенты с генетическим заболеванием могут иметь редкие или сверхредкие мутации, разработка персонализированной терапии может оказаться чрезвычайно сложной задачей. Нынешний путь регулирования ставит существенный барьер в виде требования обширных доклинических исследований и высокой стоимости товара, который может иметь неопределенное прогностическое значение. Возможно, новые пути регулирования могли бы стать катализатором разработки дополнительных методов генотерапии, в частности, для редких показаний, которые могли бы сохранить акцент на безопасности, обеспечивая при этом доступ пациентов к инновационным и сложным методам лечения. Как только терапия попадает на клинические испытания, пациенты обычно должны будут соответствовать строгим критериям включения/исключения, поэтому экстраполировать применимость новых методов терапии к различным типам пациентов, в том числе с более тяжелыми заболеваниями, может быть сложно. После одобрения терапии высокая стоимость и сложность препаратов могут стать проблемой для доступа пациентов и масштабируемости. При прочих равных условиях более дешевые и простые методы лечения были бы более доступны для пациентов.
|