Посещений: 
С CNG-КАНАЛАМИ СВЯЗАННЫЕ РЕТИНОПАТИИ
Патобиология и генотерапия
Biology, Pathobiology and Gene Therapy of CNG Channel-Related Retinopathies Maximilian J. Gerhardt, Siegfried G. Priglinger,Martin Biel
et al.
 Biomedicines Volume 11 Issue 2, 269
10.3390/biomedicines11020269
|
The visual process begins with the absorption of photons by photopigments of cone and rod photoreceptors in the retina. In this process, the signal is first amplified by a cyclic guanosine monophosphate (cGMP)-based signaling cascade and then converted into an electrical signal by cyclic nucleotide-gated (CNG) channels. CNG channels are purely ligand-gated channels whose activity can be controlled by cGMP, which induces a depolarizing Na+/Ca2+ current upon binding to the channel. Structurally, CNG channels belong to the superfamily of pore-loop cation channels and share structural similarities with hyperpolarization-activated cyclic nucleotide (HCN) and voltage-gated potassium (KCN) channels. Cone and rod photoreceptors express distinct CNG channels encoded by homologous genes. Mutations in the genes encoding the rod CNG channel (CNGA1 and CNGB1) result in retinitis-pigmentosa-type blindness. Mutations in the genes encoding the cone CNG channel (CNGA3 and CNGB3) lead to achromatopsia. Here, we review the molecular properties of CNG channels and describe their physiological and pathophysiological roles in the retina. Moreover, we summarize recent activities in the field of gene therapy aimed at developing the first gene therapies for CNG channelopathies

|
Циклические нуклеотиды, такие как cAMP и cGMP, являются вторичными мессенджерами, которые регулируют важные сигнальные пути в нашем организме путем контроля активности нескольких эффекторных белков, включая циклический нуклеотид-связывающий домен (CNBD), содержащий катионные каналы. Среди CNBD-содержащих ионных каналов циклические нуклеотиды-связывающие каналы (CNG) являются единственными строго лиганд-связывающими каналами, поскольку для их открытия требуется связывание cAMP или cGMP [1]. У позвоночных животных семейство генов каналов CNG включает шесть гомологичных членов. CNGA1, CNGA2 и CNGA3 кодируют субъединицы, которые наделяют канал ключевыми свойствами и, как было показано, образуют функциональные гомотетрамерные ионные каналы в гетерологичных системах экспрессии [1]. CNGA4, CNGB1 и CNGB3 кодируют структурно сходные субъединицы, которые сами по себе не могут образовывать функциональные ионные каналы, но важны для правильной локализации нативных канальных комплексов и придают каналу специфические биофизические свойства [1]. Четыре гена каналов CNG связаны с inherited retinal disorders (IRD): известно, что мутации в CNGA1 и CNGB1 вызывают retinitis pigmentosa (RP), а мутации в CNGA3 и CNGB3 приводят к achromatopsia (ACHM).
2. Insights on Structure and Activation of CNG Channels
Каждый из шести генов каналов CNG кодирует мембранный белок с шестью α-спиральными трансмембранными сегментами (S1-S6), ядром канала, состоящим из петли reentrant pore (P) между S5 и S6, и цитозольными N- и C-концами. S5, S6 и промежуточная петля reentrant поры образуют собственно поровый домен [2] (рис. 1). Как и в классических связанных с напряжением (voltage-gated ) каналах, S4 содержит множество положительно заряженных остатков, а S1-S4 образуют voltage-sensor-like domain (VSLD). Однако, в отличие от канонических каналов, структура VSLD сегментирована, и положительно заряженные аминокислоты расположены не регулярно [3]. Это препятствует соответствующему перемещению заряда и помогает объяснить, почему функция канала CNG не зависит от напряжения [3]. С-концевой участок содержит CNBD и соединен с S6 через С-связующее звено (рис. 1). Одночастичные криоэлектронные микроскопические структуры нативных каналов CNG палочек и колбочек человека подтвердили гетеротетрамерную стехиометрию 3:1 нативного комплекса каналов палочек [4] и колбочек [5], ранее постулированную на основании биофизических и биохимических экспериментов [6-9] (рис. 1). Нативные каналы CNG в наружных сегментах палочковых фоторецепторов представляют собой гетеротетрамеры, состоящие из трех субъединиц CNGA1 и одной субъединицы CNGB1. Канал CNG в наружных сегментах фоторецепторов колбочек образован тремя субъединицами CNGA3 и одной субъединицей CNGB3. В отличие от других членов суперсемейства эл. напряжение-генерирующих каналов, но подобно HCN-каналам, четыре субъединицы тетрамерного комплекса канала CNG расположены в не-переключаемой конфигурации, где VSLD взаимодействует только с поровым доменом той же субъединицы [3,10].
 Figure 1. span class=ari8>Structure and activation of CNG channels. (A) Membrane topology of CNG channel subunits: 1-6, transmembrane segment 1-6; C, carboxy-terminus; CNBD, cyclic nucleotide-binding domain; N, amino-terminus. (B) Model of the CNG channel complex embedded in the plasma membrane based on the human rod CNGA1/CNGB1 channel structure (PDB 7RHH). (C) Top and bottom views of the heterotetrameric human rod CNGA1/CNGB1 channel complex. (D) Subunit composition of the CNG channels from rods and cones. Structures in this figure were generated with the RSCB PDB 3D View tool (www.rcsb.org/3d-view/ accessed on 19 Dec 2022) based on PDB 7RHH.
Структуры канала CNG, полученные в различных состояниях активации в присутствии cGMP и/или фармакологических блокаторов, выявили детали архитектуры ионпроводящей поры и внесли вклад в наше понимание функции канала и влияния патогенных мутаций на функцию канала [4,5,11-14]. На основании сравнения имеющихся структур в открытом и закрытом состояниях и предыдущих исследований мутагенеза [15-18] считается, что активация канала CNG включает скоординированные движения по крайней мере трех основных элементов: CNBD, С-связывающего звена с его пропускающим (gating) кольцом и ворот канала [10,19]. Связывание циклических нуклеотидов с CNBD приводит к вращательному изменению всего С-конца относительно поры. С-линкер, домен, который аллостерически обеспечивает связывание циклических нуклеотидов с воротами канала через свое gating кольцо, также следует за этим вращением и частично перемещается вверх. Ворота канала, расположенные во внутриклеточной части сегмента S6, сужаются и, предположительно, удерживаются в закрытом состоянии постоянными силами С-линкера. После связывания лиганда описанные выше движения вызывают ослабление ингибирующих сил С-связывающего звена, и пора канала открывается, позволяя ионам проникать внутрь. Несмотря на то, что идентичность последовательностей составляет всего около 35%, структуры CNGB1 и CNGA1 хорошо выравниваются и демонстрируют сходное расположение доменов, что приводит к довольно симметричной поре закрытого гетеротетрамерного канала CNGA1/CNGB1 [4]. В гомотетрамерных каналах CNG все четыре субъединицы А демонстрируют симметричные вращательные движения, которые приводят к открытию поры. Однако открытие гетеротетрамерного канала CNGA1/CNGB1 происходит асимметрично [4]. Только две субъединицы CNGA1 слева и напротив субъединицы CNGB1 демонстрируют движения, известные для гомомерного канала, тогда как CNGB1 и другая субъединица CNGA1 почти не двигаются, что приводит к асимметричной геометрии открытой поры [4]. Структуры гетеромерного конусного канала CNG в различных состояниях активации до сих пор неизвестны. Однако анализы структуры выявили асимметричную архитектуру поры уже в закрытом состоянии с аргининовым остатком S6 CNGB1, выступающим непосредственно в поре и перекрывающим путь ионной проводимости поры [5,14]. Мутация этого аргинина на глицин привела к увеличению проводимости одиночного канала [4]. Несмотря на эти значительные успехи в понимании структуры и функции CNG-каналов, в наших знаниях все еще есть пробелы, особенно в отношении механизма стробирования (пропускной способности) CNG-канала колбочек или N- и С-концевых областей, которые по техническим причинам пока удалось смоделировать только для CNG-канала палочки быка [4,5,14]. Дальнейшее понимание ожидается от исследований, направленных на определение структуры специфических патогенных мутантов канала CNG, вызывающих ACHM или RP, с пока необъяснимыми эффектами на структуру и функцию канала.
3. Role of CNG Channels in Signal Transduction in Photoreceptors
У позвоночных имеется два типа высокоспециализированных фоторецепторов, палочки и колбочки, которые имеют сходные, но различающиеся сигнальные каскады фототрансдукции и они позволяют распознавать свет в различных условиях окружающей среды (рис. 2). Палочки обеспечивают зрение при слабом освещении, в то время как при дневном свете зрение обеспечивается колбочками и лишь в меньшей степени палочками. Колбочковая зрительная система также обеспечивает цветовое зрение, поскольку она может различать длины волн, сравнивая входные сигналы от двух (у большинства позвоночных) или трех (у человека и некоторых не-человекообразных приматов) типов колбочек, которые оснащены различными вариантами опсина колбочек с различной спектральной чувствительностью [20,21]. Как в палочках, так и в колбочках передача сигнала происходит по одному и тому же принципу и облегчается ферментами, которые контролируют концентрацию циклического гуанозинмонофосфата (cGMP). В свою очередь, cGMP контролирует активацию канала CNG в плазматической мембране наружных сегментов (рис. 2). В темноте постоянная активность трансмембранных гуанилилциклаз приводит к высоким концентрациям cGMPs, которые поддерживают каналы CNG в открытой конформации [22,23]. CNG-каналы проводят постоянный, неинактивирующий Na + и Ca 2+ ток ("темновой ток"), который деполяризует фоторецептор и способствует высвобождению глутамата в синаптических окончаниях фоторецептора. В ответ на конформационное изменение, вызванное светом, опсины, будучи рецепторами с G-белковой связью, высвобождают свой G-белок трансдуцин, который, в свою очередь, связывается с фосфодиэстеразами для cGMP типа PDE6 и активирует их [24]. Ферменты PDE6 гидролизуют cGMP, что приводит к закрытию канала CNG и гипеRPоляризации фоторецептора, тем самым уменьшая синаптическое высвобождение глутамата. Приток Ca 2+ в наружные сегменты опосредован исключительно каналами CNG [1,25,26] и сбалансирован оттоком Ca 2+ через Na +/Ca 2+, K + обменники [25,27-30]. Закрытие каналов CNG при световой стимуляции, наряду с постоянной активностью Na +/Ca 2+, K + обменников, приводит к снижению внутриклеточной концентрации Ca 2+. Это снижение концентрации Ca 2+ способствует восстановлению после световой реакции путем модуляции активности PDE6 и гуанилилциклазы [24,31-33].
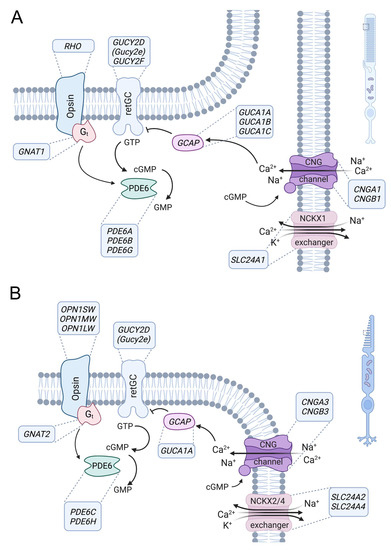 Figure 2. Role of CNG channels in rod and cone photoreceptor signaling. Phototransduction in outer segments of rod (A) and cone (B) photoreceptors. The principle of the phototransduction is similar in both cell types, but key proteins are encoded by distinct but homologous genes (human gene names are indicated in the boxes next to the proteins). In the dark, the cyclic nucleotide-gated (CNG) channel (CNGA1/B1 in rods and CNGA3/B3 in cones) of the outer membrane is kept open by high concentrations of cyclic guanosine monophosphate (cGMP) produced by retinal guanylyl cyclase (retGC) (note: GUCY2E is a pseudogene in humans, whereas Gucy2e is functional in rodents and the major retGC encoding gene). The resulting influx of Na+ and Ca2+ depolarizes the plasma membrane. Light activates the opsin, which in turn activates transducin (Gt), whose alpha subunit activates a phosphodiesterase (PDE6) that leads to hydrolysis of cGMP. The decrease in the cGMP concentration leads to the closure of the CNG channel, resulting in membrane hyperpolarization. Ca2+ is an important regulator of phototransduction. At high concentrations, Ca2+ binds to guanylyl-cyclase-activating proteins (GCAP), leading to the inhibition of retGC. High Ca2+ concentrations also lead to a slight reduction in the cGMP affinity of the CNG channel through Ca2+/calmodulin-mediated feedback inhibition (not illustrated). Ca2+ is cleared from the outer segment via a Na+-Ca2+-K+-exchanger (NCKX1 in rods, NCKX2/4 in cones). At low Ca2+ levels, Ca2+-free GCAPs can bind and activate retGC to stimulate cGMP production and reopen the CNG channel.
Ключевые действия в этом каскаде фототрансдукции одинаковы в палочках и колбочках, но часто опосредуются функционально эквивалентными белками, кодируемыми разными генами. Это также относится к каналам CNG палочек и колбочек, которые имеют практически одинаковые функциональные свойства и отличаются только некоторыми специфическими особенностями. Яркими примерами являются более высокая Ca2+ проницаемость колбочкового CNG-канала и более сильное Ca2+-зависимое ингибирование чувствительности к лигандам в канале палочкового CNG [1,34]. Однако эти различия не могут полностью объяснить разную чувствительность и кинетику палочек и колбочек [1,35].
4. Genetics and Biology of the Rod and Cone CNG Channel
4.1. The Rod CNGA1/CNGB1 Channel
4 Дисфункция канала CNG палочки вызывает аутосомно-рецессивный пигментный ретинит (RP) [36,37]. RP представляет собой генетически разнообразную группу прогрессирующих дегенеративных заболеваний сетчатки, поражающих в основном фоторецепторы сетчатки [38]. Общие симптомы RP включают ночную слепоту, прогрессирующее сужение поля зрения, аномальную миграцию и накопление пигмента в сетчатке [39]. Заболевание характеризуется первичной потерей функции палочек с последующей дегенерацией и потерей палочковых фоторецепторов, которая может варьироваться от пациента к пациенту в зависимости от основной генной мутации. По мере прогрессирования потери палочек нарушается морфология и функция колбочковых фоторецепторов, что влияет на дневное зрение. Это приводит к постепенному сужению поля зрения при дневном свете. На поздних стадиях RP приводит к ухудшению остроты зрения, а на последней стадии может привести к слепоте. Более 70 генов уже связаны с RP, с различными формами наследования [40] (см. также https://web.sph.uth.edu/RetNet/, доступ 19 декабря 2022 года). Многие гены RP кодируют белки, участвующие в каскаде фототрансдукции (Рисунок 2) или белки, необходимые для поддержания архитектуры фоторецепторов. CNGA1 (OMIM #123825) и CNGB1 (OMIM #600724) связаны с аутосомно-рецессивным RP (arRP). Распространенность мутаций, вызывающих CNGA1-RP, варьирует в разных географических регионах в пределах 1-8% случаев arRP [37,38,41-44]. Большинство выявленных мутаций CNGA1 вызывают делеции ключевых функциональных доменов или приводят к нарушению транспортировки мембранами [1,2,37,45]. На мутации в CNGB1 приходится 1-4% случаев arRP, и, как ожидается, их распространенность будет варьироваться в разных географических областях [36,38,40,42,46-49]. Хотя известные мутации CNGB1 вызывают лишь незначительные делеции или единичные аминокислотные замены, фенотип сравним с фенотипом RP у пациентов с CNGA1-RP. Были проведены исследования функциональной характеристики некоторых мутаций CNGB1, которые показали, что некоторые мутации влияют на стабильность или транспорт стержневого канала CNG, тогда как другие не влияют на экспрессию, но приводят к возникновению функционально неактивных каналов CNG [36,48,50,51].
Сегодня существуют животные модели как для CNGA1-RP, так и для CNGB1-RP [52-58] (Таблица 1). Однако большинство фенотипических данных получено на животных моделях Cngb1, некоторые из которых существуют уже более двух десятилетий и подробно описаны [54-62].
Естественно встречающаяся мутация Cnga1 была выявлена у породы шетлендских овчарок с прогрессирующей атрофией сетчатки [52]. Однако подробная информация о фенотипе сетчатки до сих пор отсутствует. Недавно появилась модель мыши с направленной делецией в экзоне 2 Cnga1 [53]. Нокаутные мыши Cnga1 несут делецию 65 п.н. со сдвигом рамки считывания, которая, хотя и не подтверждена экспериментально на уровне белка, должна приводить к преждевременному стоп-кодону и потере большей части белка Cnga1 вскоре после делеции. У гомозиготных мышей наблюдается потеря большинства фоторецепторов в возрасте 16 недель [53]. Адаптированные к темноте ответы электроретинограммы (ЭРГ) на одиночную вспышку 3 кд*с/м2 были значительно снижены у этих мышей через 3 недели, и еще больше снизились через 10 недель [53]. Совсем недавно была создана и охарактеризована модель мышей-мутантов Cnga1, вызванная N-этил-N-нитрозомочевиной (ENU) [63]. Мутантные мыши несут мутацию c.1526 A → G в Cnga1, которая приводит к обмену Y509C в CNBD белка Cnga1. Y509 соответствует Y513 в белке CNGA1 человека и участвует в формировании b3-полосы CNBD [1]. Мутация Y509C, по-видимому, нарушает стабильность комплекса канала CNG палочки, что приводит к полной потере белков Cnga1 и Cngb1, несмотря на практически неизменные уровни мРНК. В результате к трехнедельному возрасту ЭРГ-ответы, управляемые палочками, были ослаблены. Не функционирующие палочки со временем дегенерировали, и с шестого месяца жизни наблюдалась вторичная прогрессирующая дегенерация колбочек, которая завершилась к возрасту 1 год, когда ЭРГ-ответы уже не поддавались измерению [63].
Почти два десятилетия назад была описана первая модель мыши с нокаутом Cngb1 [54]. Эта модель мыши несет делецию экзона 26 гена Cngb1, который кодирует S6. Делеция также приводит к сдвигу рамки считывания, который создает стоп-кодон в первом триплете экзона 27, тем самым прекращая трансляцию [54]. Это приводит к потере экспрессии белка Cngb1, а также к деградации белка Cnga1, который, по-видимому, требует Cngb1 для правильной экспрессии. У таких мышей с нокаутом Cngb1-X26 полностью отсутствует функция канала CNG палочек, что проявляется в снижении реакции палочек на свет и уменьшении scotopic ЭРГ. Дисфункция сопровождается прогрессирующей дегенерацией палочек и вторичной дегенерацией преимущественно незатронутых колбочек. Дегенерация колбочковых фоторецепторов начинается в возрасте 6 месяцев, когда потеряно около 50% палочек. В возрасте около 1 года в сетчатке остается только 10-20% фоторецепторов [54]. В целом, фенотипы Cngb1- и Cnga1-дефицитных мышей очень похожи в плане проявления и прогрессирования заболевания. Скорее всего, это связано с тем, что потеря одной из субъединиц канала CNG приводит к вторичной деградации и потере оставшегося белка субъединицы канала CNG. В обоих случаях это приводит к полной потере комплекса стержневого канала CNG и его функций, что объясняет фенотипическое сходство, наблюдаемое в соответствующих моделях мышей.
Спонтанная мутация в CNGB1 была также обнаружена у собак породы папильон с выраженным снижением или отсутствием функции палочек и медленно прогрессирующей дегенерацией сетчатки [55]. Интересно, что мутация c.2387delA;2389_2390insAGCTAC, обнаруженная в этой естественно встречающейся модели собак, приводит к преждевременному прекращению синтеза белка Cngb1 почти в том же положении, что и в сконструированных мышах Cngb1-X26 [54,55,58]. Сравнительный анализ показал, что пациенты с CNGB1-RP и мышиные и собачьи модели с дефицитом Cngb1 имеют сходный фенотип, характеризующийся ранней потерей функции палочек и медленной дегенерацией палочковых фоторецепторов наряду с вторичным снижением функции колбочек [59]. Существование этой собачьей модели для CNGB1-RP имеет большое значение для оценки трансляционного потенциала будущих генотерапий. Это связано со структурным сходством глаз собаки и человека в плане размера и, в некоторой степени, в плане пространственного распределения подтипов фоторецепторов (например, глаз собаки имеет богатую колбочками зрительную полоску, которая частично имитирует макулу человека). Более того, тот факт, что мутанты CNGB1 у собак и Cngb1-X26 у мышей имеют генетические изменения, которые приводят к прекращению синтеза транскрипта в почти одинаковом положении, повышает уверенность в экстраполяции результатов, полученных на этих двух моделях, на ситуацию у человека.
Локус CNGB1 кодирует несколько транскриптов с различными паттернами экспрессии. Самый длинный транскрипт использует все 33 экзона и производит субъединицу CNGB1 (также называемую CNGB1a) канала CNG палочки [64]. Первые 11 или 16 экзонов (включая уникальный альтернативный экзон) [64] дают начало отдельным цитозольным белкам, соответствующим участкам glutamic-acid-rich protein (GARP) в N-концевой части CNGB1a. Для изучения эффекта делеции GARP была создана другая модель мыши с генетической модификацией в экзоне 1 Cngb1 (мыши Cngb1-X1) [56]. В принципе, у мышей Cngb1-X1 наблюдались такие же функциональные дефекты, как и у мышей Cngb1-X26, но у них была более резко нарушена морфология наружного сегмента палочки, что позволяет предположить, что растворимые и связанные с каналами белки GARP необходимы для морфогенеза дисков палочки и целостности наружного сегмента. Дополнительные исследования показали, что прикрепленные к каналу и растворимые GARPs по своей природе являются развернутыми [65] и играют различные роли в формировании морфологии наружного сегмента палочки, транспорта и функции каналов палочки CNG [50,61,64,66-69]. Эти результаты актуальны для пациентов с CNGB1-RP, которые имеют мутации в части локуса CNGB1, дающей начало белкам GARP.
4.2. The Cone CNGA3/CNGB3 Channel
Дисфункция канала CNG колбочек вызывает ахроматопсию (ACHM), редкое заболевание сетчатки, которое наследуется по аутосомно-рецессивному типу и поражает примерно одного из 30 000 человек [70]. В отличие от дальтонизма, при котором мутации в генах, кодирующих различные фотопигменты колбочек, влияют только на спектральную чувствительность [71], ACHM имеет серьезные последствия для всех аспектов дневного зрения. Симптомы включают плохую остроту зрения, фотофобию, нистагм и отсутствие цветовой дискриминации [72]. Эти симптомы отражают функциональный дефект конусных фоторецепторов, который возникает в раннем младенчестве и характеризуется отсутствием адаптированной к свету ЭРГ, но с сохранением scotopic сигнала ЭРГ [73,74]. В дополнение к функциональным нарушениям, в богатой колбочками центральной части сетчатки могут наблюдаться структурные изменения - от потери колбочек наружных сегментов до глубокой атрофии сетчатки [72]. До 90% случаев ACHM обусловлены мутациями в CNGA3 (OMIM #216900) и CNGB3 (OMIM #262300) [75-77]. Остальные случаи обусловлены мутациями в ATF6 (OMIM #616517), GNAT2 (OMIM #613856), PDE6C (OMIM #613093), PDE6H (OMIM #610024) или пока неизвестных генах [78,79].
На сегодняшний день установлено, что более 250 мутаций в CNGA3 [80-88] и более 160 мутаций в CNGB3 [76,77,80,83,89-94] вызывают ACHM у человека. Мутации в CNGB3 чаще встречаются в Европе и США и составляют 50-60% случаев ACHM [76,89], а в Нидерландах даже почти 90% [83]. Большинство мутаций CNGB3 - это нонсенс, сдвиг рамки считывания или сплайс-мутации [77,80]. Миссенс-мутация в гене CNGB3 (S435F) была выявлена у дальтоников, происходящих с атолла Пингелап в Микронезии [90]. На этом небольшом острове ACHM встречается очень часто и затрагивает почти 10% коренного населения [90-97]. По оценкам, 28-36% пациентов в западной популяции несут мутации в CNGA3 (ACHM2) [81,84]. В популяциях Ближнего Востока и Китая мутации в CNGA3 составляют около 80% случаев ACHM [42,80,98,99]. Интересно отметить, что в подгруппе пациентов с АХМН была обнаружена дигенная и триаллельная модель наследования с мутациями в CNGA3 и CNGB3 [100]. Большинство мутаций CNGA3 являются миссенс-мутациями, затрагивающими только отдельные аминокислотные остатки белка [81-88]. Предполагается, что нарушается сворачивание, внутриклеточные преобразования и транспорт [101]. Несмотря на то, что были получены некоторые сведения, точные механизмы, связывающие конкретные аминокислотные замены с фенотипом ACHM, все еще плохо изучены. Влияние отдельных аминокислотных замен на функцию белка CNGA3 изучалось в основном in vitro [85,100-116].
Существует несколько генетически модифицированных и естественных животных моделей ACHM (Таблица 1), которые помогли выяснить механизмы заболевания и служат в качестве моделей болезни для доклинической разработки новых генотерапий [58,70]. Первой описанной животной моделью ACHM была нокаутная мышь Cnga3, которая несет гомозиготную делецию экзона 7, что приводит к удалению всех доменов канала вниз по течению от третьего трансмембранного сегмента (S3), включая пору и CNBD [117]. Это приводит к полной потере функции конусного канала CNG. Как следствие, у мышей с нокаутом Cnga3 с рождения наблюдается селективный дефицит колбочковых световых реакций [117], а затем прогрессирующая дегенерация и гибель клеток колбочек [117,118]. Дегенерация колбочек затрагивает М- и S-конусы по-разному, и гибель клеток происходит значительно быстрее в вентральной и назальной (богатой S-конусами), чем в дорсальной и височной (богатой М-конусами) частях сетчатки. Вентральные колбочки почти полностью исчезают после третьего постнатального месяца, тогда как остаточные дорсальные колбочки присутствуют даже у старых нокаутных мышей [118]. Кроме того, была описана естественная мышиная модель ACHM. Линия мышей, обозначенная cpfl5 (cone photoreceptor function loss 5), несет точечную мутацию в Cnga3, приводящую к замене p.T203A в цитоплазматической петле между S2 и S3. До настоящего времени не было зарегистрировано ни одной миссенс-мутации, затрагивающей этот филогенетически консервативный треонин, соответствующий p.T224 в белке CNGA3 человека. Хотя точный механизм остается неясным, эта мутация приводит к потере экспрессии белка Cnga3 и фенотипу, аналогичному тому, который наблюдается у мыши-нокаута Cnga3 [119].
Более того, сообщалось о естественной модели ACHM у овец с врожденным нарушением зрения, характеризующимся снижением функции колбочек, но нормальной функцией палочек [120,121]. Было установлено, что пораженные ягнята были гомозиготными по нонсенс-мутации в Cnga3 (p.R236X) [121]. Впоследствии у другой породы была выявлена миссенс-мутация Cnga3 (с.G540S), вызывающая аналогичный фенотип дневной слепоты [120]. Кроме того, были описаны две спонтанные собачьи модели ACHM [122]. Одна из них - лабрадор-ретривер с мутацией p.V644del, которая удаляет консервативный валин в С-концевом домене, имеющем важное значение для сборки и стабильности гетеротетрамерного канала [6]. Вторая модель - немецкая овчарка, несущая мутацию p.R424W. Эта аминокислота находится в gating кольце внутри С-линкера, соединяющего трансмембранный домен S6 с CNBD, и является консервативной у эукариот. Важно отметить, что мутация, приводящая к замене R на W в соответствующей последовательности CNGA3 человека (p.R410W), также была обнаружена у пациентов с ахроматопсией [82]. Недавние исследования криоэлектронной микроскопии (крио-ЭМ) с каналом C. elegans версии tax-4 (CNGA3/p.R421W) пролили свет на потенциальный патогенный механизм этой миссенс-мутации [11]. Тщательный анализ данных крио-ЭМ в сочетании с электрофизиологическими и биохимическими данными привел к выводу, что эта замена R на W в gating кольце дестабилизирует закрытое состояние канала и способствует спонтанному открытию канала в отсутствие лиганда [11]. Таким образом, если канал экспрессируется в наружных сегментах колбочек пациентов с CNGA3/p.R421W, его чрезмерная активность может вызывать (например, Са2+-опосредованную) гибель клеток и дегенерацию колбочек.
Существуют также различные животные модели для CNGB3-связанной ACHM (Таблица 1). Была описана нокаутные мыши Cngb3 с генетической делецией, которая вызывает сдвиг рамки считывания и удаляет часть S1 и все остальные домены канала [123]. У этих нокаутных мышей отсутствует экспрессия белка Cngb3 и сильно снижен уровень белка Cnga3. Отсутствие канала CNG в колбочках серьезно нарушает функцию колбочек и приводит к прогрессирующей дегенерации колбочек, напоминающей фенотип нокаутных мышей Cnga3 [123,124]. В этой модели обнаруживается остаточная функция колбочек, которая, скорее всего, обеспечивается нерегулярными гомомерными каналами CNGA3. В дополнение к нокаутной мыши Cngb3 была описана естественная модель мыши, обозначенная cpfl10 [125]. Мыши несут точечную мутацию c.692G→A, приводящую к p.R231H [125]. Базовая характеристика линии мышей выявила потерю ЭРГ-ответов, управляемых колбочками, и медленную прогрессирующую дегенерацию колбочек [125].
Две естественно встречающиеся модели дегенерации колбочек у собак с мутациями в Cngb3 были идентифицированы у пород аляскинский маламут и немецкий короткошерстный пойнтер [126]. Генетический анализ показал, что у аляскинского маламута весь ген удален, а у немецких короткошерстных пойнтеров заболевание вызвано миссенс-мутацией c.784G→ A;p.D262N, затрагивающей консервативный остаток аспартата в S2 [126]. У пораженных щенков аляскинского маламута развивается дневная слепота и фотофобия, напоминающие клинический фенотип пациентов с ACHM. Симптомы проявляются только при ярком свете, в то время как зрение при тусклом свете нормальное. Сигналы конусной ЭРГ начинают уменьшаться через несколько недель после рождения и исчезают у старых собак [127]. Недавно стратегия CRISPR-Cas9 с использованием вирусного вектора была использована для создания модели нокаута in situ CNGB3-ACHM у циномольгусовых обезьян [128]. Эта остро протекающая модель может предоставить ценную информацию о патобиологии дефицита CNGB3 в сетчатке не-человекообразных приматов с фовео-макулярной структурой, аналогичной человеческому глазу.
Существование многочисленных животных моделей CNGA3- и CNGB3-ACHM не только способствует лучшему пониманию биологии канала CNG в колбочках, но и значительно облегчает разработку и тестирование потенциальных методов лечения. Как уже упоминалось для CNG-канала палочек, наличие моделей крупных животных с морфологическим сходством с человеческим глазом (по размеру и распределению клеток) имеет первостепенное значение для трансляционных исследований. Кроме моделей CNG-каналов палочек , Cnga3- и Cngb3-дефицитные мышиные модели демонстрируют некоторые фенотипические различия, возможно, из-за специфических для колбочек морфологических особенностей, которые в отсутствие Cngb3 все еще обеспечивают довольно эффективный транспорт гомотетрамерных каналов Cnga3 в наружный сегмент, тем самым поддерживая остаточную функцию CNG-канала. Однако в отсутствие Cnga3 Cngb3 не может поддерживать функцию канала самостоятельно.
Таблица 1. Обзор генов CNG сетчатки, связанных с ними заболеваний человека, моделей животных и доклинических исследований. NCT ID, идентификатор www.clinicaltrials.gov (доступ получен 19 декабря 2022 года). OMIM, Online Mendelian Inheritance in Man. POC, POC, prove of concept. 5. Gene Therapy for the Treatment of CNG Channelopathies
На сегодняшний день не существует лечения ни одной каналопатии CNG, и клиническое лечение в настоящее время ограничивается специализированной генетической консультацией, использованием вспомогательных средств для зрения и тонированных контактных линз или очков для уменьшения симптомов фотофобии. Улучшение понимания биологии каналов CNG, доступность подходящих животных моделей и появление эффективных и безопасных адено-ассоциированных вирусных векторов (AAV) привели к началу нескольких программ генотерапии для потенциального лечения ретинопатий, связанных с каналами CNG (Таблица 1).
ACHM и RP, вызванные мутациями в генах CNG-каналов, наследуются по аутосомно-рецессивному типу. В отличие от большинства аутосомно-наследственных ретинопатий, здесь нет необходимости удалять (доминантный) патогенный вариант, и для терапевтического эффекта достаточно добавить функциональную копию гена. Поэтому подходы генотерапии направлены на добавление здоровой копии вызывающего заболевание гена в пораженные клетки (в данном случае в колбочковые или палочковые фоторецепторы). Некоторые из этих так называемых подходов по дополнению (или увеличению) генов уже достигли клинической стадии развития.
5.1. The AAV Vector Technology
AAV - это маленькие (диаметр 25 нм), неразвивающиеся, непатогенные ДНК-вирусы, которые могут реплицироваться только в присутствии адено, папилломо или герпеса вирусов. Существует множество встречающихся в природе серотипов AAV, которые были исследованы на предмет использования в качестве векторов для переноса генов [149,150]. Платформа векторов AAV уже прошла клиническую оценку, и за последние годы было одобрено пять продуктов генотерапии по офтальмологическим, ЦНС и другим показаниям (www.ema.europa.eu и www.fda.gov accessed on 19 December 2022). Более трех десятилетий назад AAV были векторизованы путем замены вирусных генов rep и cap на кассету экспрессии генов по выбору [151]. Гены rep и cap AAV передаются способом транс в процессе производства рекомбинантных AAV в виде вспомогательных плазмид [152]. AAV состоят из 60-мерного капсида из структурных вирусных белков (VP1, VP2 и VP3), собранных в соотношении 5:5:50, и одноцепочечного ДНК-генома длиной около 4,7 кб, содержащего кассету для экспрессии интересующего гена, фланкированную двумя инвертированными терминальными повторами (ITR) [153]. Такие вирусные векторы AAV могут доставлять свой геном в ядро клетки-мишени, где он остается эписомным и транскрибирует интересующий ген без интеграции в геном хозяина. В последние годы AAV-векторы стали золотым стандартом доставки генов в фоторецепторы сетчатки и пигментные эпителиальные клетки сетчатки с доказанным тропизмом и эффективностью. Их успех основан на том, что они легко производятся в больших масштабах и демонстрируют в целом хороший профиль безопасности с ограниченной иммуногенностью и дозозависимой токсичностью [154,155].
5.2. Gene Therapy for CNG-Channel-Linked RP
На сегодняшний день не существует подходов генотерапии для CNGA1-связанного RP. Для CNGB1-связанного RP были проведены успешные исследования по доказательству концепции генного дополнения на основе AAV в модели мыши с нокаутом Cngb1 [129] и модели собаки с мутацией Cngb1 [59]. Чтобы обеспечить эффективную упаковку и специфическую для палочек экспрессию относительно большой полноразмерной кДНК Cngb1 (~4 кб), в этих двух исследованиях использовалась кассета экспрессии AAV с коротким промотором, специфичным для палочек [129] или фоторецептора [59], чтобы стимулировать экспрессию соответствующей виду кДНК Cngb1 (например, мыши или собаки). У обоих видов субретинальная инъекция терапевтических векторов с дополнением нормального гена с AAV (серотип 5 или 8) приводила к эффективной экспрессии белка Cngb1 и восстановлению экспрессии и локализации канала CNG. Это привело к улучшению опосредованной палочками функции сетчатки, сохранению структуры сетчатки и задержке вторичной дегенерации колбочек. Наконец, мыши с нокаутом Cngb1, а также собаки с мутацией CNGB1, прошедшие лечение, показали значительно лучшие результаты, чем контрольные особи без лечения, в тестах на поведение, управляемое зрением, зависящим от палочек [59,129]. Эти многообещающие результаты способствовали началу трансляционных исследований с использованием гуманизированной версии вектора (AAV5-RHO-CNGB1), в котором короткий промотор человеческого родопсина стимулирует экспрессию полноразмерного человеческого CNGB1 [130]. При введении однократной субретинальнойю инъекции 4-недельным мышам с нокаутом Cngb1, AAV5-RHO-CNGB1 привел к эффективной экспрессии человеческого белка CNGB1 в мышиных палочках и восстановил экспрессию эндогенного мышиного белка Cngb1 [130]. Лечение привело к дозо-зависимому восстановлению ЭРГ-ответов, управляемых палочками, и к сохранению структуры сетчатки [130]. В настоящее время проводятся исследования на крупных животных моделях, чтобы поддержать применение этого подхода генотерапии для будущего лечения пациентов с CNGB1-RP.
5.3. Gene Therapy for CNG-Channel-Linked ACHM
5.3.1. ACHM Gene Therapy: Preclinical Proof-of-Concept Studies
Вектор AAV5, экспрессирующий кДНК CNGB3 человека под контролем одной из трех различных усеченных версий промотора опсина M/L человека, был оценен в методе увеличения генов у собак, страдающих ахроматопсией из-за мутаций в CNGB3 [143]. Собакам делали односторонние инъекции в субретинальное пространство в возрасте от 3 до 81 недели. Улучшение функции колбочек наблюдалось уже через 4 недели после лечения в фотопических условиях с помощью ЭРГ и поведения и сохранялось не менее 14 месяцев. Наилучшие результаты лечения были достигнуты у животных 3-недельного возраста, тогда как у собак в возрасте 1 года и старше лечение было минимально эффективным [143]. Точные причины зависимости лечения от возраста неизвестны, но они могут быть связаны с морфологическими изменениями, наблюдаемыми на более поздних стадиях заболевания. Соответственно, эффективность лечения в этой модели была повышена у пожилых собак, когда увеличение генов AAV сочеталось с введением цилиарного нейротрофического фактора (CNTF), который, как известно, вызывает временную деконструкцию наружных сегментов фоторецепторов [144].
Положительное доказательство концепции генотерапии с использованием вектора AAV5, обеспечивающего экспрессию кДНК Cnga3 мыши под контролем короткого промотора S-опсина мыши, было также достигнуто в модели ахроматопсии с нокаутом Cnga3 [132]. У двухнедельных мышей, которых лечили с помощью субретинальной инъекции, наблюдались колбочковые ЭРГ-ответы, нормализация уровня cGMP и экспрессии комплексов колбочковых CNG-каналов и опсинов, а также задержка гибели колбочковых клеток. Кроме того, ганглиозные клетки из обработанных, но не обработанных мышей с нокаутом Cnga3 демонстрировали вызванную светом пиковую активность, что свидетельствует о том, что сигналы, генерируемые в наружной сетчатке, передаются в мозг. Наконец, было продемонстрировано, что вновь полученная сенсорная информация преобразуется в поведение, управляемое зрением с помощью колбочек [132]. Терапевтический эффект был стабильным в течение как минимум 12 месяцев и также наблюдался при использовании вектора серотипа AAV8 или при лечении в возрасте 3 месяцев [133]. Аналогичные эффекты были получены в различных моделях мышей Cnga3 после субретинального введения вектора AAV5 и промотора человеческого опсина M/L [119], а также интравитреальной доставки сконструированных векторов AAV8 (Y447, 733F) [134] или AAV2.GL [135]. В соответствии с исследованиями на собаках, вектор AAV8, обеспечивающий экспрессию кДНК CNGB3 человека под контролем короткого промотора ARR3 человека, был показан как эффективное средство для восстановления ЭРГ реакции колбочек и остроты зрения в мышиной модели ахроматопсии с нокаутом Cngb3 [145]. Успешная терапия с использованием AAV-генов также была описана на модели ахроматопсии CNGA3 у овец Awassi [136]. Значительное долгосрочное улучшение функции колбочек было продемонстрировано в течение как минимум 6 лет после однократного введения AAV-вектора, экспрессирующего человеческий CNGA3 [137,138].
Эти многообещающие доклинические исследования привели к началу реализации в общей сложности пяти независимых программ генотерапии ахроматопсии, связанной с CNGA3 и CNGB3. Исследования безопасности на овцах и не-человекообразных приматах выявили некоторое воспаление после субретинальной доставки генов CNGA3, но в целом показали приемлемый профиль безопасности по крайней мере для двух различных используемых продуктов генотерапии CNGA3 [138-140,156]. Данные по безопасности третьего продукта генотерапии CNGA3 пока не опубликованы. Для одной из двух программ CNGB3 были опубликованы данные по безопасности, полученные на мышах, собаках и циномольгусовых обезьянах, которые показали приемлемую безопасность в отношении воспаления и токсичности, зависящих от вектора и дозы [146-148]. 5.3.2. ACHM Gene Therapy: Clinical Studies Все вышеупомянутые трансляционные программы для CNGA3- и CNGB3-связанной ахроматопсии уже достигли клинической фазы [70] (Таблица 1). Немецкий академический исследовательский консорциум RD-CURE инициировал первое клиническое исследование, в котором оценивался эффект трех различных доз (1 x 1010, 5 x 1010 и 1 x 1011 геномов вектора на глаз в целом) AAV8. CNGA3, введенных посредством субретинальной инъекции в один глаз. В исследовании приняли участие девять пациентов в трех группах доз. Несмотря на высоко инвазивную процедуру введения, которая включала витрэктомию и отслойку фовео-макулярной сетчатки, лечение хорошо переносилось и приводило к независимым от дозы легким и преходящим побочным явлениям, связанным с процедурой или лекарственными препаратами [157,158] и преходящей субклинической индукции маркеров воспаления [140,142]. Лечение привело к улучшению вторичных end points, связанных с функцией колбочек, включая улучшение остроты зрения и контрастной чувствительности по сравнению с исходным уровнем у всех пациентов после лечения [158], которое имело дозо-зависимую тенденцию и сохранялось по крайней мере до 3 лет после лечения [157]. В настоящее время продолжается клиническое исследование фазы IIb, направленное на лечение второго глаза первых пациентов и лечение детей в возрасте от 6 до 12 лет (Таблица 1).
Четыре другие программы в настоящее время находятся в фазе I/II клинических испытаний, две из них по CNGA3-ACHM и еще две по CNGB3-ACHM (Таблица 1). Предварительные данные по безопасности и эффективности клинических испытаний, спонсируемых промышленностью, в которых проверялась безопасность и эффективность продуктов генотерапии AGTC-401 и AGTC-402 при ахроматопсии, связанной с CNGB3 и CNGA3, соответственно, были представлены на ежегодной встрече Американского общества по исследованию зрения и офтальмологии (ARVO). Оба продукта генотерапии использовали модифицированные капсиды AAV2 с тремя поверхностными мутациями Y-F (Y275F, Y447F и Y733F), обозначенный AAV2tYF, и промотор человеческого опсина M/L размером 1,7 кб, стимулирующий экспрессию либо CNGA3, либо CNGB3. В исследование AGTC-401 были включены 21 взрослый и 10 педиатрических пациентов с ахроматопсией CNGB3 в шести группах доз (1,2 х 1011 векторных геномов (vg)/mL до 3,2 х 1012 vg/mL). Исследование AGTC-402 включало 16 взрослых и 8 педиатрических пациентов с ахроматопсией CNGA3, распределенных на пять дозовых групп (от 4 x 1010 vg/mL до 3,2 x 1012 vg/mL). В обоих исследованиях у детей при самой высокой дозе (3,2 x 1012 vg/mL) была отмечена токсичность, ограничивающая дозу, которая включала увеит и изменения в заднем сегменте. У взрослых пациентов самая высокая доза переносилась лучше. Генотерапия улучшила светочувствительность у некоторых пациентов с CNGB3-ACHM, но в меньшей степени у пациентов с CNGA3-ACHM. Для обеих программ были начаты долгосрочные последующие исследования (Таблица 1), но недавно компания объявила, что программа CNGA3-ACHM не будет развиваться дальше. Другие спонсируемые промышленностью программы генотерапии CNGA3 и CNGB3 опубликовали лишь ограниченные данные о безопасности на сайте clinicaltrials.gov (https://clinicaltrials.gov/ct2/show/results/NCT03758404, доступ получен 19 декабря 2022 года), но сообщили о планах начать клинические исследования на поздних стадиях.
|