Посещений: 
СЛУХОВЫЕ ГЕНЕТИЧЕСКИЕ НАРУШЕНИЯ
Генотерапия
Current Advances in Gene Therapies of Genetic Auditory Neuropathy Spectrum Disorder Anissa Rym Saidia, Jerome Ruel, Amel Bahloul et al.
 J. Clin. Med. 2023, 12(3), 738; https://doi.org/10.3390/jcm12030738
|
Auditory neuropathy spectrum disorder (ANSD) refers to a range of hearing impairments characterized by an impaired transmission of sound from the cochlea to the brain. This defect can be due to a lesion or defect in the inner hair cell (IHC), IHC ribbon synapse (e.g., pre-synaptic release of glutamate), postsynaptic terminals of the spiral ganglion neurons, or demyelination and axonal loss within the auditory nerve. To date, the only clinical treatment options for ANSD are hearing aids and cochlear implantation. However, despite the advances in hearing-aid and cochlear-implant technologies, the quality of perceived sound still cannot match that of the normal ear. Recent advanced genetic diagnostics and clinical audiology made it possible to identify the precise site of a lesion and to characterize the specific disease mechanisms of ANSD, thus bringing renewed hope to the treatment or prevention of auditory neurodegeneration. Moreover, genetic routes involving the replacement or corrective editing of mutant sequences or defected genes to repair damaged cells for the future restoration of hearing in deaf people are showing promise. In this review, we provide an update on recent discoveries in the molecular pathophysiology of genetic lesions, auditory synaptopathy and neuropathy, and gene-therapy research towards hearing restoration in rodent models and in clinical trials.

|
Слух у млекопитающих зависит от способности сенсорных волосковых клеток преобразовывать вызванные звуком механические стимулы в электрохимические сигналы. Отклонение пучка волосков вызывает быстрое открытие каналов сенсорной трансдукции, что приводит к притоку катионов в IHC. Это приводит к деполяризации потенциала, обеспечивая приток кальция через зависимые от напряжение (вольтаж) кальциевые каналы. Соединение Ca 2+ каналов в пресинаптическом участке ленточного (ribbon ) синапса вызывает высокоскоростное слияние синаптических везикул и высвобождение нейромедиатора глутамата из синаптической щели. Высвобождение глутамата в синапсе активирует чувствительные r Ca 2+ AMPA-рецепторы (рис. 1). Это инициирует генерацию нейронных шипиков (spikes) в волокнах спирального ганглионарного нейрона (SGN), который кодирует информацию о звуковых стимулах, передаваемую в центральную нервную систему. Дисфункция на любом уровне этого сложного механизма трансдукции может нарушить кодирование акустических характеристик, особенно временных сигналов. Потенциальные места повреждений разнообразны, включая IHCs ленточные синапсы IHCs или синаптопатию (например, пресинаптическое высвобождение глутамата или нарушения постсинаптических окончаний дендритов нейронов спирального ганглия), или могут быть вызваны демиелинизацией и потерей аксонов слухового нерва и их мишеней в кохлеарном ядре (т.е. невропатией, рис. 1). Эти слуховые патологии получили название расстройства спектра слуховой нейропатии (ANSD), при которых активность наружных волосковых клеток (OHCs) сохраняется (рис. 1) [1,2,3,4].
 Figure 1. Inner hair cell (IHC)-spiral ganglion synaptic complex. The IHC is connected to all type I spiral ganglion neurons (SGNs) forming the radial afferent system (red) going to the cochlear nuclei. The OHC synapses with small endings from type II spiral ganglion neurons, forming the spiral afferent system (green). Molecular composition of a mature ribbon of the inner hair-cell synaptic complex ensuring the temporal precision of peripheral sound encoding. The mature ribbon synapse between the sensory inner hair cells (IHCs) and postsynaptic spiral ganglion neurons (SGNs) involves the spatial confinement of several molecular components: presynaptic density merge to one single ribbon anchor and, postsynaptically, one continuous elongated postsynaptic density composed by functional synaptic AMPA-preferring glutamate receptors, but also some silent NMDA receptors. Font color indicates association with the correspondingly colored pre-/postsynaptic localization.
Клинические характеристики ANSD весьма неоднородны в зависимости от разнообразия этиологии. ANSD может быть результатом синдромальных и несиндромальных генетических аномалий, а также экологических причин (например, гипоксия, шумовое воздействие, цитотоксические онкологические препараты) и старения. ANSD является одной из распространенных причин потери слуха, поражая от 1,2% до 10% людей с потерей слуха [5]. Аудиологические ANSD характеризуется легкой или глубокой нейро-сенсорной тугоухостью, с нарушением или отсутствием сложных потенциалов действия (CAP) и слуховых реакций ствола мозга (ABRs, рис. 2) и ухудшением речевой аудиометрии в тишине [6]; они связаны с нормальной отоакустической эмиссией (OAE, рис. 2) или кохлеарной микрофонией (CM), что указывает на нормальную функцию OHCs. Кроме того, обычно наблюдается отсутствие стремянного рефлекса среднего уха и контралатеральное подавление отоакустической эмиссии [1,5,7,8].
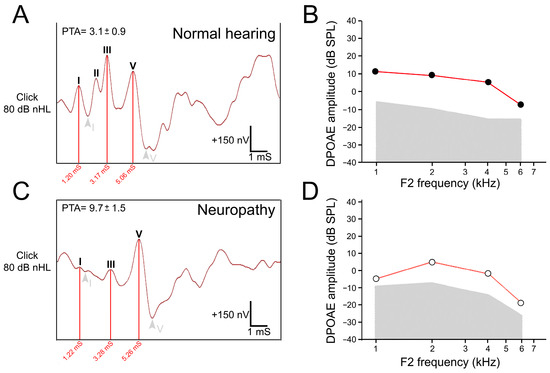 Figure 2. Auditory brainstem responses (ABRs) and distortion product of otoacoustic emissions (DPOAEs) recorded from one normal hearing control (A,B) and one patient with speech understanding difficulties (C,D). (A) ABRs were recorded at 80 dB nHL (decibels normal hearing level) with normal amplitudes, latencies, and morphology. (B) No abnormalities were detected in the DPOAEs. (C) A patient with speech understanding difficulties showed a clear decrease in the amplitude of waves I, II, and III. The I-V interval was 4.04 vs. 3.86 for the patient and a normal hearing subject, respectively. (D) DPOAEs were still present in this patient. PTAs: mean of pure-tone audiometry thresholds recorded at 250, 500, 1000, 2000, 3000, and 4000 Hz. Note that PTA is within the normal range in the patient with speech understanding difficulties. The audiologic results of this patient suggest a hidden auditory neuropathy.
Электрокохлеография (ECochG) и тесты нейронной адаптации остаются мощным диагностическим инструментом, помогающим определить место поражения. Например, отсутствие суммирующего потенциала на ECochG указывает на потерю механоэлектрической трансдукции IHCs или самих IHCs. Кроме того, ECochG можно использовать для отличия слуховой синаптопатии от слуховой нейропатии. Действительно, пациенты со слуховой синаптопатией демонстрировали повышенную адаптацию к частотно-специфическим звукам [9]. Напротив, пациенты со слуховой нейропатией демонстрировали нормальную адаптацию к низкочастотным звукам, но аномально повышенную адаптацию к высокочастотным звукам [9]. У пациентов со слуховой синаптопатией аномальная адаптация к громкости, вероятно, связана с нарушением, затрагивающим IHC-ленточные синапсы в Кортиевом органе вдоль базилярной мембраны [9]. В случаях слуховой нейропатии потеря нервных волокон равномерно распределена по всей улитке, и поэтому наблюдаемое нарушение нейронной проводимости, вероятно, не зависит от происхождения волокон вдоль базилярной мембраны [2]. Поэтому нормальная адаптация, наблюдаемая в низкочастотной области, является неожиданной и может отражать компенсацию со стороны центральных слуховых структур, участвующих в восприятии громкости, уменьшая импульсы слухового нерва специфическим для частоты образом [10,11].
Hidden hearing loss (HHL) или supraliminal нарушения слуха, вероятно, являются специфическим типом ANSD , вызванным, например, воздействием шума, старением или периферической нейропатией, и характеризуются нормальными порогами чистого слуха в сочетании с дефицитом вызванной звуком активности слухового нерва (рис. 2). Пациенты с HHL демонстрируют нормальные пороги речевой аудиометрии в спокойных, хорошо синхронизированных ABR, но с нарушением распознавания речи в шумной обстановке [12].
В настоящее время клиническими вариантами восстановления слуха у пациентов, страдающих от ANSD , являются слуховые аппараты, которые могут усиливать звук при легкой или умеренной глухоте, или кохлеарные имплантаты при тяжелой глухоте [12]. Преимущество последних заключается в том, что они могут обходить нефункционирующие сенсорные волосковые клетки, напрямую стимулируя остаточные слуховые нейронные структуры в глухой улитке. На самом деле, опубликованные в литературе данные о долгосрочных результатах реабилитации слуха с помощью слуховых аппаратов у детей с ANSD частично противоречивы [13-15]. Некоторые сообщают о хорошей реабилитации слуха [13-15], в то время как другие описывают отсутствие слуховой и коммуникативной пользы от слуховых аппаратов у детей с ANSD [1,16]. Кохлеарные имплантаты, которые электронно стимулируют SGNs могут обеспечить эффективную слуховую реабилитацию для пациентов со слуховой синаптопатией, поскольку генерация и распространение шипов (spikes) поддерживается. Как правило, пациенты с поражением слухового нерва плохо работают с кохлеарными имплантами, что, вероятно, связано с изменением нейронной передачи электрического сигнала от кохлеарного импланта [12]. Однако у пациентов с нейропатией слухового нерва, связанной с OPA1, через год после имплантации наблюдалось улучшение восприятия речи и синхронная активация слуховых путей [17], вероятно, в обход места поражения (которое может быть расположено в терминальных дендритах) и/или благодаря электрическим стимулам, вызывающим четко выраженную временную активацию SGNs. Однако на сегодняшний день данные о результатах реабилитации слуховой нейропатии ограничены [5,18].
За последнее десятилетие произошел значительный прогресс в понимании молекулярных патогенетических механизмов, которые способствуют развитию нарушений слуха, вызванных экологическими и генетическими факторами. Это, в свою очередь, дало новую надежду концепциям замены или исправления мутантных последовательностей или дефектных генов с целью предотвращения слуховой нейродегенерации или содействия регенерации слуховых синапсов и нервных волокон.
2. Pathogenic Mechanisms of Auditory Neuropathy
Синдромная слуховая нейропатия поражает множество черепных и периферических нервов, в то время как несиндромные слуховые нейропатии ограничиваются слуховым нервом. Большинство случаев несиндромальной слуховой нейропатии является результатом нарушения синаптической передачи [5].
2.1. Non-Syndromic Auditory Synaptopathies
Генетические слуховые синаптопатии обычно вызывают только глухоту, например, мутации в гене CACNA1D, кодирующем Ca2+ канал Cav1.3L-типа, гене OTOF, кодирующем Otoferlin, гене SLC17A8, кодирующем Vglut3, или гене DIAPH3, кодирующем диафановый формин 3.
2.1.1. Otoferlin-DFNB9
Ген OTOF кодирует отоферлин, который является важнейшим кальциевым сенсором для синаптического экзоцитоза в кохлеарных IHCs [19,20]. Отоферлин также участвует в реформации везикул, в повторном снабжении и связывании в активной зоне, что делает отоферлин многоцелевым белком [20,21]. Мутации в гене, кодирующем отоферлин, ответственны за аутосомно-рецессивную глубокую прелингвальную глухоту, DFNB9 [22]. На сегодняшний день выявлено около 220 патогенных вариантов в гене OTOF [23]. Предполагается, что большинство этих мутаций являются нонсенс-мутациями или укорочениями, которые провоцируют инактивацию отоферлина [24]. У пациентов с вариантами в OTOF наблюдалась более легкая потеря слуха, а также прогрессирующая и чувствительная к температуре потеря слуха, в то время как OAEs были сохранены [22,25-27]. У детей с биаллельными мутациями гена OTOF наблюдалась глубокая потеря слуха, отсутствие ABR и CAP, но сохранение DPOAEs и амплитуды CM [28]. Мыши с нокаутом гена Otoferlin, которые глубоко глухи из-за сбоя вызванного звуком высвобождения нейротрансмиттера в синапсах IHC, вероятно, будут подходящей животной моделью для DFNB9 [29,30]. У этих мышей Ca2+-триггерный экзоцитоз в IHCs почти отсутствует [29,30]; синаптические везикулы были обнаружены рядом с мембраной в активной зоне, что говорит о том, что отсутствие везикул не ограничивает передачу сигнала, а нарушает поздний этап экзоцитоза [30].
2.1.2. VGLUT3-DFNA25
Везикулярные транспортеры глутамата (VGLUTs) отвечают за загрузку глутамата в синаптические везикулы, что необходимо для осуществления синаптической передачи [31]. VGLUT3 экспрессируется в небольших подмножествах нейронов в центральной нервной системе [31,32]. У мышей VGLUT3 экспрессируется в IHCs [33,34] и OHCs [35]. Генетическое устранение Slc17a8 у мышей приводит к отсутствию эффектов CAP или ABR на акустические стимулы, в то время как ABR могут быть вызваны электрическими стимулами, и у этих мышей была зарегистрирована сильная отоакустическая эмиссия [33,34]. Таким образом, это отражает нарушение активации восходящего слухового пути, в то время как активность в OHCs не нарушена [33-37]. У пациентов с делецией 12q22-q24 в гене SLC17A8 в локусе DFNA25 наблюдается врожденная и несиндромальная аутосомно-доминантная глухота [33,38,39]. Глухота у пациентов характеризовалась как высокочастотная, прогрессирующая нейросенсорная тугоухость с хорошим восстановлением слуха с помощью кохлеарной имплантации, что подтверждает гипотезу о синаптопатии [33,39]. У мышей VGLUT3A224V/A224V, несущих мутацию p.A221V (p.A221V у людей соответствует p.A224V у мышей) в гене Slc17a8, наблюдалась прогрессирующая потеря слуха при неповрежденной функции OHC [40]. Однако суммирующий потенциал был снижен, что указывает на изменение рецепторного потенциала IHC. Сканирующая электронная микроскопия показала разрушение пучков стереоцилий IHC, при этом пучки стереоцилий OHC остались незатронутыми. Кроме того, ленточные синапсы IHC претерпевали структурные и функциональные изменения на более поздних стадиях. Эти результаты позволяют предположить, что DFNA25 возникает в результате сбоя механотрансдукции с последующим изменением синаптической передачи [40].
2.1.3. Cav1.3-SANDD
Приток кальция в основание IHCs вблизи ленточного синапса опосредуется через кальциевый канал L-типа (Ca2+) Cav1.3, который является основным вольтаж-сопряженным Ca2+ каналом в IHCs и необходим для слуха. Cav1.3 преобразует вызванную звуком деполяризацию в высвобождение нейротрансмиттера глутамата в синаптическом участке, что приводит к передаче сигнала на слуховой нерв [41]. Cav1.3, кодируемый геном CACNA1D, широко распространен среди различных клеток, таких как OHCs, IHCs, кардиомиоциты, нейроэндокринные клетки и нейроны. Мутация Cav1.3 в CACNA1D может вызывать дисфункцию синоатриального узла и глухоту (так называемый синдром SANDD) у мышей и у людей, причем у людей, близко напоминающую таковую у мышей Cacna1d-/- [41,42]. Cav1.3 необходим для нормального слуха и сердечного ритма у людей, и потери функции только подмножества каналов достаточно, чтобы вызвать синдром SANDD [42]. Мутации с потерей функции в гене CACNA1D вызывают нарушение синаптической нейротрансмиссии в ленточном синапсе IHC у нокаутных мышей [41,43]. Cav1.3 защищает сенсорные волосковые клетки во время старения улитки путем снижения кальций-опосредованного окислительного стресса у мышей-самцов C57BL/6J [44] и играет важную роль в дифференцировке внутреннего уха [45].
2.1.4. CABP2-DFNB93
Кальций-связывающий белок 2 (CABP2) является мощным модулятором IHC вольтаж-связанных кальциевых каналов CaV1.3. CABP2 регулирует приток Ca2+ в пресинаптическом участке [46,47] и, таким образом, везикулярный выброс глутамата. Патологические мутации в CABP2 приводят к аутосомно-рецессивному, умеренно-тяжелому несиндромальному нарушению слуха DFNB93 [48-51]. Пациенты с DFNB93 демонстрируют фенотип слуховой синаптопатии с нормальными OAEs [52]. Используя нокаутную модель мыши, Picher et al. [52] продемонстрировали, что нарушение слуха при DFNB93 может быть результатом усиленной устойчивой инактивации каналов CaV1.3 в синапсах IHCs, что ограничивает их доступность для запуска синаптической передачи, приводя к повышению слуховых порогов [52]. Это, однако, не мешает развитию улитки и не вызывает ранней дегенерации волосковых клеток или их синаптического комплекса [52,53]. Эти результаты свидетельствуют о расширенном окне для генотерапии.
2.1.5. DIAPH3-AUNA1
Слуховая нейропатия, несиндромальная, аутосомно-доминантная 1 (AUNA1) - это форма прогрессирующей глухоты человека с поздним началом, возникающая в результате точечной мутации в нетранслируемой области 5 гена Diaphanous homolog 3 (DIAPH3). Мутация DIAPH3 приводит к избытояной экспрессии белка DIAPH3, члена семейства форминов, участвующего в динамике цитоскелета [54]. У пациентов с AUNA1 отсутствует или изменен ABR, в то время как функции OHC сохраняются [1,55], что указывает на слуховую нейропатию. У трансгенных мышей, избыточно экспрессирующих Diap3, наблюдается прогрессирующее смещение порога, но сохраняется продукт искажения отоакустической эмиссии (DPOAEs) [54,56]. Морфологическая оценка выявила избирательное и рано начинающееся изменение кутикулярной пластинки IHC и слияние стереоцилий с последующей потерей способности IHC передавать входящие сенсорные стимулы [54,56]. Кроме того, в течение 24 недель у мутантных мышей наблюдалось значительное снижение числа ленточных синапсов IHC, хотя это снижение не коррелировало по времени с началом и прогрессированием потери слуха или аномалий пучка стереоцилий [54]. В совокупности эти результаты позволяют предположить важную функцию Diap3 в регуляции сборки и/или поддержания актиновых филаментов в стереоцилиях IHC, а также потенциальную роль в ленточных синапсах IHC.
2.2. Syndromic Auditory Neuropathy
Генетические нейропатии часто поражают разные нейроны, что приводит к синдромным фенотипам, таким как болезнь Charcot-Marie-Tooth, аутосомно-доминантная атрофия зрительного нерва, наследственная нейропатия зрительного нерва Leber's, атаксия Friedreich's, синдром Mohr-Tranebjaerg , болезнь Refsum или синдром Wolfram [57-60].
2.2.1. Charcot-Marie-Tooth
Аутосомно-доминантная болезнь Charcot-Marie-Tooth (CMT) является наиболее распространенной наследственной периферической полинейропатией, характеризующейся дегенерацией периферических нервов. CMT можно разделить на две основные категории: CMT типа 1 (демиелинизирующие невропатии) и типа 2 (аксональная форма невропатий) [61,62]. Пациенты с CMT несут мутации в генах MPZ для нулевого белка миелина или PMP22, кодирующих белки, необходимые для формирования и адгезии миелина [2,63,64]. CMT типа 1 А (CMT1А) является преобладающим подтипом, который представляет собой демиелинизирующую периферическую невропатию, характеризующуюся дистальной мышечной слабостью, потерей чувствительности, арефлексией и медленной скоростью проведения моторных и сенсорных нервов [1,62,63]. Нарушение слуха также является относительно распространенным симптомом CMT1А. По сравнению с контрольной группой, у пациентов с CMT1А значительно снижена способность воспринимать речь в шумной обстановке, а также снижено временное и спектральное разрешение, что позволяет предположить, что демиелинизация слуховых нервных волокон при CMT1А вызывает дефект кохлеарной нейротрансмиссии [65]. У пациентов с CMT 1 и 2 типа отмечается задержка или снижение амплитуды ABR, а также ухудшение разборчивости речи, что является электрофизиологическим свидетельством слуховой нейропатии [62].
2.2.2. Autosomal-Dominant Optic Atrophy
Аутосомно-доминантная атрофия зрительного нерва (DOA) является наиболее частой формой наследственной нейропатии зрительного нерва [66] с частотой 1:10 000 и вызывается гетерозиготными вариантами в гене OPA1, кодирующем митохондриальную динамин-связанную большую ГТФазу [67-69]. OPA1 участвует во многих функциях митохондрий, в частности, в поддержании дыхательной цепи и потенциала клеточной мембраны [70-72], организации крист, контроле апоптоза [72,73] и поддержании митохондриальной ДНК [74-76]. Изначально DOA была описана как несиндромальная умеренная или тяжелая потеря остроты зрения с коварным началом в раннем детстве, вызванная прогрессирующей потерей ганглиозных клеток сетчатки [77]. В последнее десятилетие клинический спектр DOA был расширен до широкого спектра симптомов, включая глухоту, атаксию, невропатию и миопатию, и теперь называется доминантной атрофией зрительного нерва плюс (DOA плюс) [74,78,79]. Глухота является вторым по распространенности клиническим признаком при DOAplus, поражая около 20% всех пациентов с DOA [17,74,78-80].
Ассоциация DOA и глухоты классически связана с мутацией R445H в экзоне 14, но в литературе уже сообщалось и о других вариантах неправильного смыслообразования OPA1 [79,81]. В этом случае потеря слуха начинается в детстве или раннем взрослом возрасте [79,82]. Хотя в большинстве исследований нарушение слуха в целом квалифицируется как "нейросенсорная тугоухость", некоторые авторы предложили в качестве патофизиологического механизма, лежащего в основе нарушения слуха при OPA1-DOA, слуховую нейропатию [17,70,83,84]. Аудиологическое обследование пациентов с нарушением слуха, имеющих миссенс-мутации OPA1, показало нарушение восприятия речи, отсутствие или значительное изменение ABR, но сохранение OAE и даже усиление СМ-потенциалов, отражающих нормальную функцию OHC [28].
2.2.3. Leber Hereditary Optic Neuropathy
Наследственная зрительная нейропатия Лебера (LHON) является наиболее распространенным митохондриальным генетическим заболеванием. Оно характеризуется двусторонней, подострой, безболезненной потерей зрения, и более 95% случаев LHON вызваны одной из трех точечных мутаций митохондриальной ДНК (мтДНК): 3460G>A, 11778G>A и 14484T>C или мутацией в гене TMEM126A, кодирующем митохондриальный белок. Тяжелая аксональная дегенерация с демиелинизацией зрительного нерва была показана при гистологическом исследовании [85]. У пациентов с наследственной оптической нейропатией Лебера также наблюдаются признаки слуховой нейропатии [86,87].
2.2.4. Friedreich's Ataxia
Атаксия Фридрейха (FRDA) является наиболее частой аутосомно-рецессивной наследственной атаксией, вызванной мутациями в гене FXN, кодирующем митохондриальный белок Frataxin, участвующий в регуляции накопления железа в митохондриях. FRDA обусловлена аномальным повторением триплета GAA (от 100 до 2000 триплетов GAA) в гене FXN [88]. Помимо нарушения равновесия и координации волевых движений, атаксия Фридрейха связана с нарушением слуха, включая трудности понимания речи при фоновом шуме; слуховые пороги, однако, не изменяются [88-91], как и функция OHCs [92]. В результате слуховой нейропатии у большинства пациентов наблюдаются аномалии в слуховых нейронных реакциях и реакциях ствола мозга [92-94]. У 8-13% пациентов с FRDA наблюдается нейросенсорная тугоухость, выявляемая на аудиограмме чистых тонов [95].
2.2.5. Mohr-Tranebjaerg Syndrome
Синдром Мора-Транебьерга, при котором глухота ассоциируется с прогрессирующей дистонией и нарушением зрения, может быть классифицирован как неизолированная слуховая нейропатия. Действительно, наблюдение посмертных образцов показывает потерю нейронов с сохранением OHCs [96]. И в этом случае в основе этого синдрома лежат мутации (DDP1 для глухоты-дистонии) TIMM8A/DDP1, который кодирует полипептид из 97 аминокислот, расположенный в митохондриях.
3. Gene Therapies for Genetic Synaptopathies and Neuropathies
Генотерапия - это экспериментальная техника, которая использует гены для лечения или профилактики заболеваний путем введения желаемого чужеродного гена или генно-регуляторного элемента, такого как РНК-интерференция, в клетки-мишени для замены или восстановления дефектного гена [97]. В будущем генотерапия может обещать восстановление слуха при некоторых формах моногенной глухоты, когда морфология улитки сохраняется в течение периода времени, позволяющего вмешаться для восстановления слуха. Несколько вирусных векторов (например, аденовирус (Ad), адено-ассоциированный вирус (AAV), лентивирус) уже использовались для трансдукции внутреннего уха [98,99]. Последние исследования были направлены на оптимизацию векторных систем на основе AAV (Таблица 1), благодаря их эффективности в трансдукции клеток сенсорного эпителия.
3.1. Restoration of Neurotransmission in IHC Synapses
3.1.1. DFNB9
Мыши с нокаутом otoferlin , которые являются глубоко глухими из-за вызванного звуком сбоя высвобождения нейротрансмиттера в синапсах IHC, вероятно, являются подходящей животной моделью для DFNB9 [29,30]. AAV-опосредованный перенос гена, кодирующего отоферлин, технически сложен из-за ограниченной способности AAV к упаковке ДНК (~4,7 кб). Это ограничение делает невозможным упаковку больших генов, таких как Otof (кДНК ~6 кб). Чтобы преодолеть эту проблему, Reisinger et al. [100] исследовали возможность восстановления слуха с помощью AAV-опосредованного переноса гена synaptotagmin1 в волосковые клетки мыши, дефицитные по отоферлину. До четвертого постнатального дня у мыши экзоцитоз кальция зависит от синаптотагмина 1 [101], тогда как синаптотагмин1/2 не экспрессируется во взрослых IHCs [102]. Таким образом, продление экспрессии синаптотагмина 1 на более длительный срок может спасти потерю отоферлина при DFNB9. К сожалению, эта стратегия не смогла восстановить экзоцитоз, запускаемый притоком Ca2+, в IHCs мышей Otof-to [100].
Akil et al [103] адаптировали метод двойного AAV-вектора для доставки больших кДНК [104]. Они использовали два различных рекомбинантных вектора, один из которых содержал 5', а другой - 3' часть гена. Они показали, что однократная доставка этих двух векторов через мембрану круглого окна в улитку мутантных мышей Otof-/- на ст. P10, P17 или P30 восстанавливала производство полноразмерного белка и частично восстанавливала слух у глухих мышей Otof-/- [103].
Совсем недавно Rankovic et al. [105] использовали новый генно-терапевтический метод избыточно нагруженных AAVs для упаковки полноразмерного Otof. Действительно, они упаковали полноразмерный Otof в несколько встречающихся в природе, а также в недавно разработанные и высокоэффективные синтетические серотипы AAV. Используя постнатальную инъекцию AAV через мембрану круглого окна, они проверили эффективность этих перенагруженных AAV для индукции экспрессии функционального отоферлина в IHC в эксплантатах улитки мыши в культурах, а также in vivo у взрослых мышей. Они добились специфической экспрессии отоферлина в ~30% всех IHC и частичного восстановления слуха у мутантных мышей Otof-/-. Эти результаты указывают на возможность использования вектора AAV для упаковки крупных генов, таких как Otof, для восстановления функции слуха (Таблица 1, Рисунок 3).
 Figure 3. Summary diagram of the design gene therapies discussed in this review. Genetic syndromic auditory neuropathy affects multiple nerves, while non-syndromic auditory synaptopathy is limited to IHC ribbon synapses. These auditory phenotypes may result from loss of function, dominant negative effects, or gain-of-function expression mechanisms. Hearing rescue strategies may be designed to target the different levels of disease mechanisms, i.e., (i) WT gene addition to rescue loss function phenotype; (ii) gene or mRNA editing to correct dominant-negative, gain-of-function, or loss of function mutations; (iii) gene silencing to correct gain-of-function or expression; and (iv) NT3 gene or protein addition to enhance the repair and/or regeneration of auditory synapses and nerve fibers. The mutated protein shown here is the OPA1 protein involved in dominant optic atrophy (DOA). 3.1.2. DFNA25
Мутации в гене SLC17A8, кодирующем VGLUT3, вызывают аутосомно-доминантную глухоту, связанную со слуховой синаптопатией. Нулевые мыши с направленной делецией экзона 2 гена Slc17a8 демонстрировали отсутствие ABRs, вызванных акустической стимуляцией, в то время как ABRs, вызванные электрическими стимулами, были сохранены, наряду с интактными ОАЕ [33,34]. Успешное восстановление слуха было продемонстрировано в этой модели Slc17a8-нулевs[ мышей путем восстановления экспрессии Vglut3 через постнатальную AAV-опосредованную доставку, что иллюстрируется восстановлением синаптической передачи и слуха [36]. Недавнее исследование показало, что AAV8, экспрессирующий Vglut3 в слуховых улитках у 5-, 8- и 20-недельных Vglut3-нулевых мышей, привел к экзогенной экспрессии Vglut3 во всех IHC и к успешному восстановлению слуха в течение по крайней мере 12 недель посредством canalostomic инъекции AAV-Vglut3 [106] (Таблица 1).
У пациентов DFNA25 с мутациями в гене SLC17A8 [33,38,39] наблюдалась прогрессирующая нейросенсорная тугоухость на высоких частотах, которая также характеризовалась как синаптопатия [33,39]. Однако до сих пор не предпринимались попытки AAV-опосредованной передачи генов для исправления мутировавших последовательностей и восстановления слуха в моделях грызунов с DFNA25.
3.1.3. DFNB93
Было показано, что патологические мутации в гене CABP2 у человека вызывают умеренное или тяжелое, несиндромальное аутосомно-рецессивное нарушение слуха DFNB93, характеризующееся как слуховая синаптопатия [48-52]. Недавнее интересное исследование показало эффективность через круглую мембранну инъекции AAV2/1- и AAV-PHP.eB-опосредованной экспрессии CABP2 в IHCs P5-7 постнатальных Cabp-/- мышей в восстановлении функции Cav1.3 в IHCs и улучшении слуха у Cabp-/- мышей [51] (Таблица 1, Рисунок 3).
3.2. Hearing Restoration in Syndromic Auditory Neuropathy
3.2.1. Charcot-Marie-Tooth
Charcot-Marie-Tooth типа 1А (демиелинизирующие нейропатии) вызывается дупликацией гена PMP22. В результате тандемной внутрихромосомной дупликации размером 1,4 Мб на хромосоме 17p11.2-p12 образуются три копии гена, каждая из которых транслируется в белок PMP22 [61,107-109]. Генотерапевтические подходы к лечению CMT1A были разработаны для снижения избыточной экспрессии PMP22 на уровне ДНК или мРНК. С этой целью в моделях CMT1A у мышей и крыс были протестированы вектор RNA-interference (RNAi). [110] AAV2/9, экспрессирующий мышиную shRNA, нацеленную на PMP22 [111], и miR-318, снижающий уровень мРНК PMP22 [112]. Эти терапевтические подходы нормализовали уровни белков MPZ и PMP22 и улучшили миелинизацию, функцию, двигательную активность и электрофизиологические параметры [111-114]. Кроме того, подкожное введение антисмысловой, нацеленной на PMP22, shRNA крысам с CMT1A также снижало уровень мРНК Pmp22 и улучшало функциональные и морфологические нарушения в модели грызунов с CMT1A в зависимости от дозы [115]. Однако, как и в случае метода RNAi, антисмысловая терапия требует многократного дозирования. CRISPR/Cas9-опосредованная делеция промотора TATA-box гена PMP22 у мышей с помощью невирусных интраневральных инъекций также понизила уровень мРНК Pmp22 и снижала патологию нервов [116]. Однако вне-целевые эффекты подходов к редактированию генов остаются предметом озабоченности, и альтернативным подходом могут быть методы редактирования мРНК, такие как транс-сплайсинг РНК, опосредованный сплайсесомами [117].
Наконец, для лечения CMT1A было предложено добавление нейротрофина-3 (NT-3), нейротрофического фактора, имеющего решающее значение для аутокринного выживания и регенерации шванновских клеток [118]. Подкожное введение пептида NT-3 мышам nude, содержащим ксенотрансплантаты CMT1A, мышам TremblerJ с 22-точечной мутацией белка периферического миелина и пациентам с CMT1A привело к улучшению аксональной регенерации в животных моделях CMT1A и обеспечило благоприятный эффект у пациентов [119]. Впоследствии та же лаборатория показала, что инъекция кДНК NT-3, упакованной в AAV1, в мышцу может действовать как секреторный орган для широкого распространения NT-3 у мышей TremblerJ с демиелинизирующей CMT. Этот терапевтический подход повысил измеримые уровни секреции NT-3 в крови в достаточной степени, чтобы обеспечить улучшение двигательной функции, гистопатологии и электрофизиологии периферических нервов мышей TremblerJ, обработанных кДНК AAV1-NT-3 [120]. Эти исследования внутримышечной доставки rAAV1 NT-3 могут послужить образцом для других заболеваний нервов, связанных с нарушением регенерации нервов. В настоящее время AAV1, несущий кДНК NT-3 человека scAAV1.tMCK.NTF3, проходит фазу I/IIa клинических испытаний (NCT03520751) с использованием двусторонних внутримышечных инъекций у пациентов с CMT1A (Таблица 1, Рисунок 3).
Несмотря на перспективность генно-терапевтических средств, разработанных для лечения CMT1A, их влияние на слух еще не оценивалось.
3.2.2. Autosomal-Dominant Optic Atrophy
Хорошо известно, что гаплонедостаточность отвечает за изолированную DOA, в то время как доминантно-отрицательные или пагубные типы усиления функции могут быть ответственны за DOA-плюс [121]. Таким образом, повышение экспрессии OPA1 представляет собой перспективный терапевтический подход к лечению OPA1-ассоциированных заболеваний. Один из таких подходов генотерапии заключается в повышении экспрессии гена OPA1 на уровне ДНК. Для этого Sarzi et al. [122] исследовали возможность восстановления зрительной функции путем интравитреальных инъекций AAV2, несущего кДНК человеческого варианта №1 OPA1, который дает начало длинной и короткой изоформам OPA1. Их результаты показали, что AAV2-опосредованная терапия добавлением WT OPA1 может быть достаточной для предотвращения дегенерации ганглиозных клеток сетчатки, хотя и без восстановления зрительной функции [122]. Недавно Jüschke и др. [123] выявили новую мутацию OPA1, c.1065+5G>A, у пациентов с DOA. Эта мутация приводит к пропуску экзона 10 OPA1 и снижению экспрессии белка OPA1 на ~50%. Однако правильная функция OPA1 зависит от тонкого баланса различных изоформ L- и S-OPA1. Авторы предложили перспективную стратегию преобразования транскриптов OPA1 с неправильным сплайсингом в транскрипты OPA1 с правильным сплайсингом и, таким образом, увеличения доли функциональных транскриптов OPA1 без изменения процессинга изоформ. С этой целью они сконструировали сплайс-факторы U1, направленные в разные места экзона 10 или интрона 10 OPA1. Они показали, что применение U1, предназначенного для связывания с интроном 10 в положении +18, привело к значительному подавлению эффекта мутации (пропуск экзона 10) и повышению уровня экспрессии нормальных транскриптов в фибробластах, полученных от пациентов с DOA [123]. Это исследование является доказательством целесообразности коррекции сплайс-мутации в качестве метода лечения DOA.
Другим потенциальным вариантом генетической терапии DOA может быть редактирование генов с помощью CRISPR-Cas9. Используя этот метод, Sladen et al. [124] успешно добились коррекции мутанта OPA1 c.1334G>A: p.R445H в 57% изолированных плюрипотентных стволовых клеток (iPSCs), полученных от пациентов с DOA. Коррекция OPA1 привела к восстановлению митохондриального гомеостаза, сетевого и базального дыхания и производства АТФ, а также к снижению восприимчивости к апоптотическим стимулам в iPSCs пациентов (Таблица 1, Рисунок 3).
В целом, эти многообещающие исследования открывают путь для изучения генотерапии слуховых функциональных изменений в мышиных моделях, несущих мутации OPA1 человека.
3.2.3. Leber Hereditary Optic Neuropathy (LHON)
Для лечения LHON были разработаны генотерапии, направленные на компенсацию дефекта митохондриального комплекса 1. Этот подход основан на доставке функционального гена WT ND4 в ядро ганглиозных клеток сетчатки и последующем импорте его в митохондрии путем добавления митохондриальной таргетной последовательности для восстановления активности дыхательной цепи [125,126]. Эта стратегия была протестирована на нескольких моделях LHON у грызунов с помощью интравитреальной доставки генов, опосредованной AAV. Различные группы показали, что интравитреальная инъекция была безопасной и не вызывала глазных осложнений, связанных с самим лечением. Они также продемонстрировали митохондриальную интернализацию AAV, экспрессию его генетического содержимого и комплементацию патогенного фенотипа [127-133] (Таблица 1, Рисунок 3).
Таблица 1. Генная терапия генетических синаптопатий и нейропатий, которые обсуждались в данном обзоре. 4. Conclusions
В данном обзоре литературы описаны патогенетические механизмы, опосредующие генетический ANSD, а также генетические методы лечения, находящиеся в стадии разработки. Открытие гена, ответственного за ANSD, открыло новое и захватывающее время для поиска лекарств и терапевтического генетического модулирования. Новые открытия продолжают стимулировать разработку инновационных методов лечения как не-синдромального, так и синдромального ANSD. В последние годы был достигнут значительный прогресс в разработке генно-терапевтических средств для регенерации слуховых синапсов и нейронов или замены дефектных генов с помощью генотерапии. Тем не менее, несмотря на прогресс в возможностях доставки генов, сложная природа процессов регенерации и восстановления и широкий спектр молекулярных и клеточных мишеней подчеркивают необходимость создания точно контролируемых систем, способных доставлять широкий спектр биоматериалов, таких как гены, siRNAs, РНК и ДНК.
Один из интересных подходов основан на использовании технологий редактирования генома на основе программируемых нуклеаз, включая CRISPR-Cas9 [134]. Эти новые технологии позволяют нам удалять или исправлять пагубные мутации или вставлять защитные мутации в больные клетки и ткани. Инъекция комплексов CRISPR-Cas9 в уши новорожденных мышей с мутацией Beethoven улучшила слуховую функцию [135], тем самым обеспечив потенциальный терапевтический вариант для глухих пациентов с моногенными мутациями. CRISPR/Cas9-опосредованная делеция промотора TATA-box гена PMP22 в мышиной модели Charcot-Marie-Tooth с помощью невирусных интраневральных инъекций также понизила уровень PMP22 мРНК и ослабила патологию нервов [116]. Однако внецелевые эффекты подходов к редактированию генов по-прежнему вызывают беспокойство.
Альтернативным подходом к исправлению генных мутаций является использование транс-сплайсинга РНК, опосредованного сплайсосомами, или SMaRT. Этот метод нацелен на последовательность мРНК для исправления мутаций. Доказательство концепции SMaRT уже было установлено в нескольких моделях генетических заболеваний, вызванных рецессивными мутациями [117]. Эта инновационная технология еще не исследовалась во внутреннем ухе, но дает надежду на единый метод лечения для восстановления слуха у пациентов с рецессивными генными мутациями.
Подход AAV-опосредованной генотерапии сделал первые шаги в 2008 году, когда была продемонстрирована эффективность генотерапии для лечения врожденного амавроза Лебера. Были завершены три успешных клинических испытания безопасности субретинальной инъекции специфического белка пигментного эпителия сетчатки 65 кДа (RPE65), экспрессированного вектором AAV, для лечения врожденного амавроза Лебера [136-139]. Опубликованные исследования клинических испытаний генов, предназначенных для компенсации дефекта митохондриального комплекса 1 функциональным геном дикого типа с помощью интравитреальной инъекции AAV2-ND4 у пациентов с ND4-LHON, сообщили о клинически значимых благоприятных эффектах, выходящих за рамки ожидаемой естественной истории болезни [125]. AAV2-ND4 успешно восстановил активность дыхательной цепи и избавил от дегенерации ганглиозных клеток сетчатки [125].
Эти испытания проложили путь к созданию первых одобренных FDA продуктов генотерапии для лечения ANSD . DB-OTO, ведущий кандидат на генную терапию, направленный компанией Decibel Therapeutics на лечение вызванного мутацией отоферлина DFNB9, получил разрешение FDA США на начало фазы 1/2 клинических испытаний на педиатрических пациентах [140]. OTOF-GT, ведущий кандидат генной терапии, разработанный компанией Sensorion biotech, также получил от Управления по контролю за продуктами и лекарствами США (FDA) статус редкого педиатрического заболевания [141] для лечения педиатрических пациентов с DFNB9. На сегодняшний день проведено множество клинических испытаний, в которых изучалась AAV-опосредованная генотерапия при нейропатиях зрительного нерва. Однако не было проведено ни одного испытания с участием слуховых нейропатий, хотя генотерапия моногенных заболеваний с помощью AAV стала возможной. Несоответствие между прогрессом генотерапии зрительных и слуховых нейропатий в основном объясняется более ранним доклиническим успехом и большей доступностью лечения глаза по сравнению с улиткой. Кроме того, гетерогенность является основной проблемой в лечении генетических ANSD, поскольку на эффективность лечения влияют несколько факторов, таких как терапевтическое окно, мишени, молекулы-мишени и функция белка.
Будущие методы лечения для восстановления синаптической передачи или регенерации волокон слухового нерва должны учитывать множество мишеней, которые учитывают сложность факторов, вызывающих заболевание, и патогенетических механизмов. Оглядываясь на исторические, функциональные и молекулярные достижения, сделанные в этой области, можно сказать, что каждое из них стало возможным благодаря технологическому развитию, определившему новую эпоху. Для того чтобы преодолеть несколько статичный текущий статус клинических испытаний, нам сейчас необходимо усовершенствовать протокол этих испытаний и искать более предсказуемые животные модели глухоты, на которых они основываются. Кроме того, необходимо разработать более надежные клинические диагностические инструменты для раннего выявления ANSD , а также тщательно оценить степень дегенерации слуховых синапсов и нервных волокон. Клинические испытания биологических агентов для лечения ANSD нуждаются в достоверных показателях клинических результатов и биомаркерах.
|